

Introduction
Glomerular basement membrane (GBM) antigens are prominent targets of abnormal immune response, which often leads to end-stage renal disease (ESRD) and consequently to dialysis or kidney transplantation. The hallmark of this immune reaction is continuous linear deposition of immunoglobulins, usually of class IgG, only in rare cases, IgA or IgM, along GBM demonstrated by immunofluorescence microscopy [1]. These findings are pathognomonic for anti-GBM disease, which is defined by the presence of serum anti-GBM antibody and/or linear binding of IgG revealed by direct immunofluorescence (DIF) in a kidney biopsy specimen [1]. The main target of anti-GBM antibodies is the non-collagenous C-terminal (NC1) domain of the α3 chain of type IV collagen (α3(IV)NC1), but in most patients that develop posttransplant anti-GBM disease, predominantly with X-linked Alport syndrome, the primary target of anti-GBM antibodies is the NC1 domain of the α5 chain of type IV collagen (α5(IV)NC1) [2, 3]. However, it should be noted that DIF of the kidney specimens in diabetic patients usually also reveals this characteristic pattern of linear homogenous staining for IgG, but the reason for that remains obscure [4].
Here we describe a case of a male patient with post-transplant anti-GBM nephritis with negative serum anti-GBM antibodies determined by indirect IF (IIF).
Case report
We observed a 33-year-old male with de novo renal failure in a kidney transplant from a deceased kidney donor. The patient had kidney transplantation two months prior to the observation and a history of ESRD diagnosed in 2013. He was maintained on usual immunosuppressive triple therapy with tacrolimus, mycophenolate and prednisone. Furthermore, there was a history of arterial hypertension, inherited thrombophilia (heterozygous factor V Leiden mutation), gastrointestinal hemorrhage, pneumonia (in 2013, successfully treated with clarithromycin), eosinophilia and erythrocyturia. He had no family history of renal disease, hearing defects or vision abnormalities. Autoimmune causes of ESRD, like lupus nephritis, granulomatosis with polyangiitis and Goodpasture syndrome were excluded before treatment was given by thorough clinical examination and laboratory tests (IIF tests for antinuclear antibodies, anti-neutrophil cytoplasmic antibodies and anti-GBM antibodies were negative).
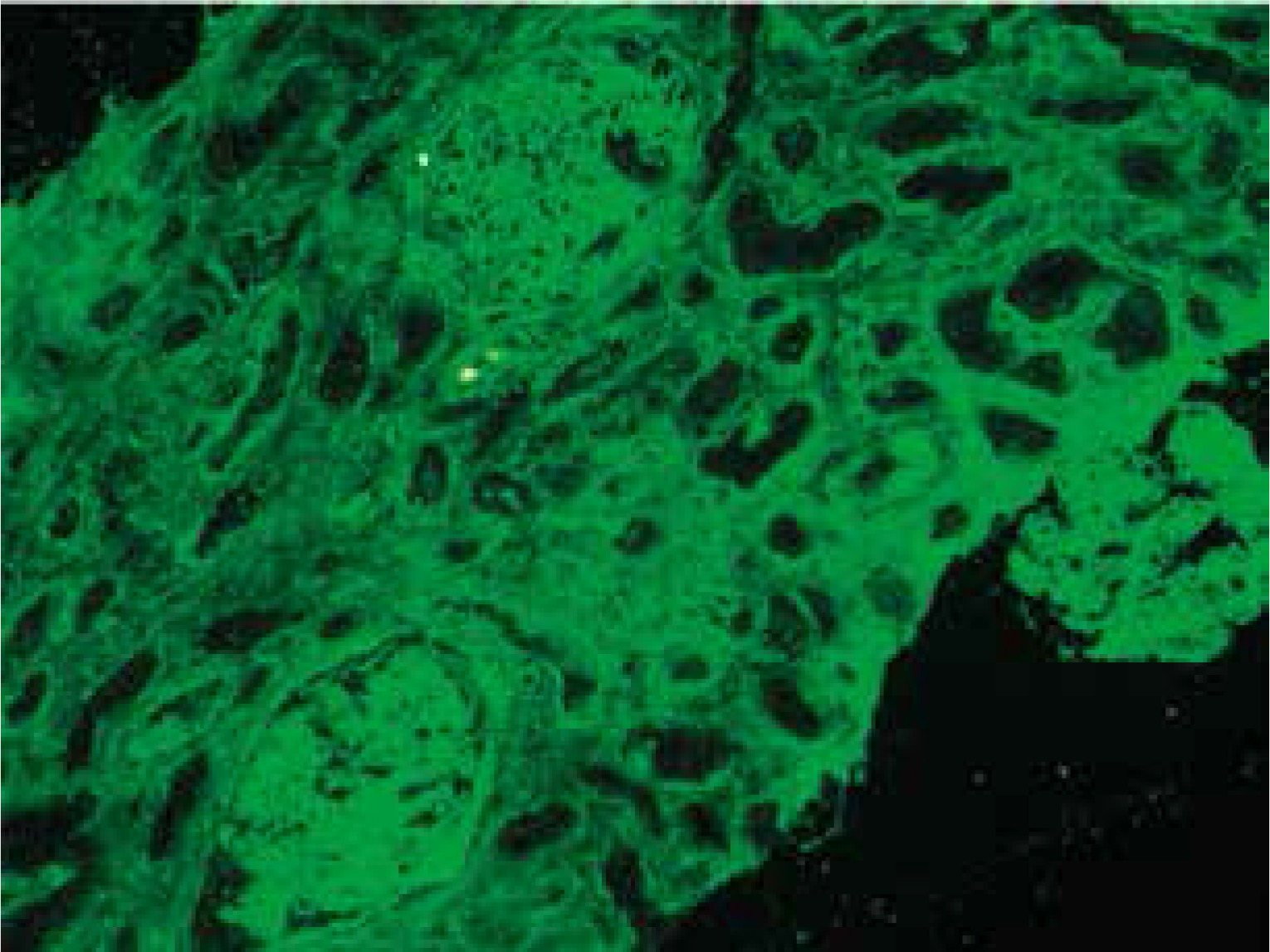
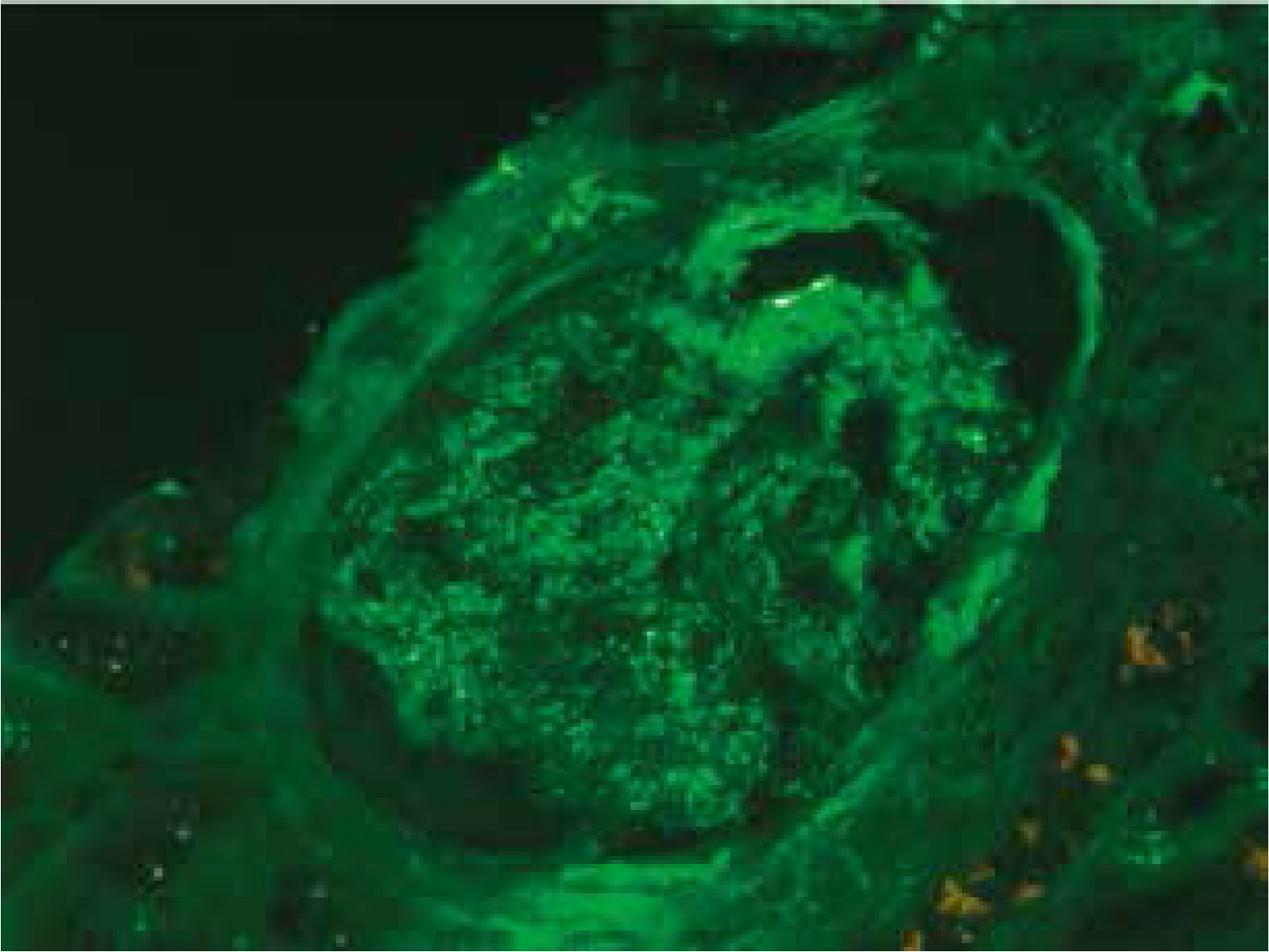
At presentation, he had no symptoms of infection. The clinical-laboratory investigations revealed increased serum levels of creatinine (2.9 mg/dl), blood urea nitrogen (82 mg/dl), normal potassium and sodium levels, low red blood cells (3.1 × 106/μl) and hemoglobin (9.2 g/dl). Protein determined in a 24-hour urine collection test was 0.1 g/24 h. The other biochemical parameters, including C-reactive protein and procalcitonin, and white blood count were unremarkable. The patient was HBsAg, anti-HCV and CMV-DNA negative. His abdomen was non-tender and abdominal ultrasound showed no signs of acute transplant rejection. The biopsy from the transplanted kidney was taken. Light microscopy showed fragments of cortex with 9-11 glomeruli, with mild mesangial hypercellularity. There was also present mild focal interstitial fibrosis and tubular atrophy with sparse lymphocyte infiltrate, which indicated rather chronic than acute rejection. Tissue was not available for electron microscopy. Immunofluorescence microscopy showed strong linear IgG staining along the capillary walls (Figs. 1 and 2) and segmental fine granular staining along the capillary walls for C3 (Fig. 3). Peritubular capillaries were negative for C4d. MHC-DR expression was also negative. Circulating anti-GBM antibodies were not detected by indirect IF. Informed consent to genetic testing was obtained. DNA was isolated from peripheral blood (QIAamp DNA Mini Blood, QIAGEN, Germany). Direct sequencing of the purified polymerase chain reaction (PCR) products for the NC1 domain of COL4A5 and COL4A3, obtained with the primers (Table 1), was performed in both directions on ABI Prism 3130 (Applied Biosystems, United States). Mutation analysis of the sequenced region of COL4A5 and COL4A3 identified no mutation, which along with the clinical picture did not confirm the diagnosis of Alport syndrome. As diabetes mellitus was excluded, the findings were interpreted as an atypical form of anti-GBM disease in the allograft.
Table 1
Oligonucleotide primers and polymerase chain reaction (PCR) conditions for the COL4A3 and COL4A5 exons analysis
[i] Primers for exons 48-51 α3(IV)NC1 were used as in [5]

Discussion
Strong linear IgG staining along GBM is diagnostic of anti-GBM disease. After renal transplantation, the incidence of linear GBM fixation is much higher in Alport syndrome patients than in patients with other renal diseases [6]. The development of anti-GBM disease in renal allograft is due to the lack of α3α4α5(IV) trimers in GBM of patients with Alport syndrome and is typically seen in approximately 4-12% of patients with this disease [7, 8]. Posttransplant anti-GBM nephritis is an allogeneic immune response to antigens absent in the native kidneys of the allograft recipient, but present in the transplanted kidney. However, in a large study of 767 renal allograft recipients, despite anti-GBM staining detected by DIF, serum anti-GBM antibodies were not detected in any patient [6]. Moreover, more than a half of patients with linear GBM staining were diagnosed with renal disease other than Alport syndrome [6]. Rendering these results into the presented case, the presence of linear GBM fixation is not evidently related to the presence of circulating anti-GBM antibodies and can be an isolated finding, and does not necessarily mean the diagnosis of Alport syndrome or diabetes mellitus. However, we cannot exclude that the titer of anti-GBM antibodies in serum were below the detectable limit. Noteworthy, the presence of linear IgG staining did not seem to considerably influence graft survival and function [6, 9], on the contrary to the presented patient.
On the other hand, although a very small portion of patients with anti-GBM nephritis lack detectable circulating anti-GBM antibodies, there are reports of presence of monoclonal anti-GBM nephritis. Recently, Coley et al. reported a case of anti-GBM nephritis with an indolent course, monoclonal IgG1κ linear staining of the GBM, but without detectable serum anti-GBM antibody in a patient initially presented with asymptomatic decreased kidney function following an upper respiratory tract infection [10]. Also Knoll et al. described a patient with anti-GBM-mediated nephritis and linear IgG3 staining, presented with hematuria and proteinuria, but normal renal function [11]. The presented case most closely resembles the case reported by Coley et al. [10], but one should note the important differences in the course of disease (stage 5 CKD and the need for kidney transplantation in the presented case, which were absent in the patient from the cited case report despite a 9-year follow-up). The limitation of the presented short communication was the method used for determining anti-GBM antibodies, which recognized all human IgG subclasses together and did not distinguish between them and the absence of immunostainings for κ/λ light chains.