
Introduction
Short stature (SS) is a significant psychosocial concern for individuals and their parents. The various etiologies of SS include normal physiological variants, such as familial short stature (FSS) and constitutional delay in growth and puberty. The pathological causes of SS are isolated growth hormone deficiency (IGHD), combined pituitary hormonal deficiency (CPHD), primary hypothyroidism, rickets, skeletal dysplasia, and various syndromic etiologies [1].
Syndromic short stature (SSS) is a clinically and genetically diverse entity with heterogeneous presentation; it is commonly associated with low birth weight and dysmorphic features but is often under-recognized and under-reported. The etiology is usually complex, involving chromosomal aberrations, copy-number variants, gene mutations, methylation defects, and unknown causes [2]. The advent of new molecular genetic techniques has revolutionized the diagnosis of SSS, enabling precise delineation of genetic etiologies, better correlation of clinical phenotypes, and informed prediction of future disease course. In addition, molecular genetic analysis helps enhance clinical management, assess prognosis, guide therapy selection, and provide genetic counseling [3]. Recombinant growth hormone (rhGH) therapy is one of the widely available growth-altering treatments to improve stature as well as psychosocial stigma.
This retrospective study highlights the various etiologies of SSS, their clinical characteristics, diverse molecular genetic variants, and rhGH treatment responses in different spectrums of syndromic short-stature children.
Material and methods
This is a retrospective study of 16 children with SSS under follow-up in the Endocrinology Department of a tertiary care referral center in South India between 2019 and 2025. Severe SS was defined as height < –3.0 standard deviations (SD), using the Indian Academy of Pediatrics (IAP) charts for age and sex [4]. SSS was diagnosed in the presence of severe SS with characteristic facial and dysmorphic features with or without intellectual disability, with confirmation by genetic testing [5]. Patients with IGHD, CPHD, idiopathic SS (ISS), chronic illness-related secondary SS, Turner’s syndrome, and skeletal dysplasia were excluded. Clinical details, including birth history, family history, parental consanguinity, and age of presentation, were recorded. All patients underwent detailed clinical examination from head to toe, with recording of all dysmorphic features and detailed anthropometry, including calculation of upper–lower segment ratios and Tanner’s staging. All cases underwent abdominal ultrasonography, cardiac evaluation, and wrist-hand X-rays for bone age assessment. Height, weight, and body mass index were recorded and plotted on the IAP growth charts at initial and follow-up visits. Assessment for developmental delay was assessed by evaluating age-dependent skills attained in the gross motor/fine motor, cognitive, language, and personal/social domains.
After ensuring normal cortisol and thyroid levels, insulin-like growth factor 1 (IGF-1) was estimated in all children, and an IGF-1 level below the third centile for age and sex was considered low. A growth hormone (GH) provocative test was performed with clonidine at a dose of 5 mcg/kg. Blood samples for GH were drawn at baseline, 60 min, and 90 min. The GH provocative test was considered normal when stimulated GH was more than 10 ng/ml, and GH deficiency (GHD) was defined when the GH level was below 7 ng/ml [6]. For intermediate responses (7–10 ng/ml), interpretation was based on auxological data, IGF-1, clinical phenotype, and imaging. All hormonal estimations were performed using the Roche Cobas e 411 ECLIA analyzer [World Health Organization International Standard: 98/574; intra-assay coefficient of variation (CV) 5.6–6.8 % and inter-assay CV of 5.8–6.6%]. A clonidine stimulation test was performed as a single provocative test according to the institutional protocol, given the severe auxological criteria, with syndromic/small-for-gestational age (SGA) background, and supportive evidence from Indian studies [7, 8].
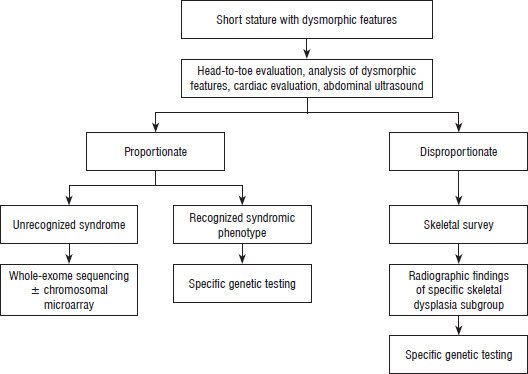
Systemic and secondary causes of growth retardation were ruled out with complete blood counts (CBC), renal function tests, IgA tissue transglutaminase (celiac disease), liver function tests, and lipid profile. Magnetic resonance imaging (MRI) of the brain was performed to evaluate the pituitary and other structural anomalies. Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) was performed in those with prenatal and postnatal growth retardation in whom Silver–Russell syndrome (SRS) was suspected. MS-MLPA was also conducted in clinically suspected cases of Prader–Willi syndrome (PWS). Subsequently, whole exome sequencing using next-generation sequencing (NGS) technology was performed in cases of unrecognized dysmorphic syndrome. Genetic testing was carried out with proper pretest counseling and informed consent (Figure 1).
RhGH therapy was administered at a dose of 0.25–0.40 mg/kg/week. The dose was adjusted based on syndrome type, growth velocity [height SD score (SDS) gain], and IGF-1 level. IGF-1 values above +2 SD prompted dose adjustments. As there is no established consensus defining an adequate height SDS response across SSS, a gain in height SDS ≥ 0.3 at 1 year was considered a positive response. All children were monitored every month for adverse effects including headache, visual symptoms, scoliosis progression, hip pain, and injection-site reactions. Anthropometry, blood pressure, fasting glucose, thyroid profile, and serum IGF-1 were assessed every 3–6 months. Scoliosis and slipped capital femoral epiphysis were screened clinically; X-rays were performed only when symptoms warranted. Fundoscopy was performed when indicated to rule out intracranial hypertension. For syndromes with increased oncogenic risk, periodic monitoring included abdominal ultrasonography and hematological evaluation (CBC with peripheral smear) every 6–12 months, with echocardiography and MRI of the brain performed annually according to the institutional protocol and syndrome-specific risk profiles. RhGH therapy was provided free of cost to all the study cohort from the Rare Disease Fund of the National Health Mission, Department of Health and Family Welfare, Government of Tamil Nadu as well as the Chief Minister’s Comprehensive Health Insurance scheme (https://www.cmchistn.com).
Ethical procedures
The study was approved by the Institutional Ethical Committee, reference No. 3374/IEC/2024-02/22. Written informed consent for the publication of clinical photographs was obtained from the parents of all 16 children.
Statistical analysis
Data normality was assessed using the Shapiro–Wilk test and visual inspection. Normally distributed variables are presented as mean ±SD; skewed data as median (IQR). Within-group pre- and post-therapy comparisons were performed using paired t-tests for normally distributed data and Wilcoxon signed-rank tests for non-normal data. Two-tailed p < 0.05 was considered significant. 95% Confidence intervals (CI) were calculated for the key outcome measure – Δ height SDS. Analyses were performed using IBM SPSS Statistics, version 27.0
Results
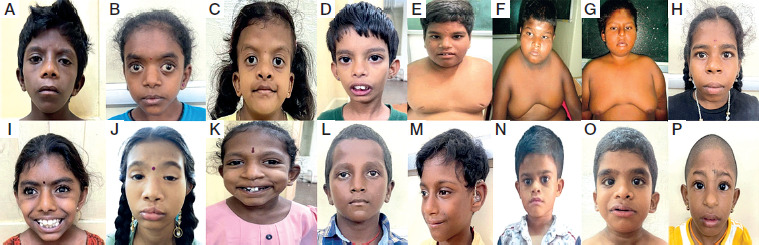
Among the 162 children with severe SS, who were on a minimum of one year of daily rhGH therapy, 18 (11.1%) were diagnosed as SSS based on dysmorphic features and genetic testing. Two children were excluded from analysis due to loss to follow-up before completing one year of therapy. Sixteen children (9.8%) completed ≥ 1 year of rhGH therapy and were included in the analysis. Among the sixteen, 8 were male and 8 were female. The mean age at presentation was 9.13 ±2.13 years, and the median baseline height SDS was –4.2 [IQR: –5.28 to –3.28], respectively. Parental consanguinity was observed in 8 (50%) of the overall cohort. Of 16 children, 9 (56.25%) were SGA or had intrauterine growth restriction (IUGR), with the overall cohort having a mean birth weight SDS of –2.33 ±1.12. In addition to SS, all had facial dysmorphism (n = 16), congenital heart disease (n = 3), intellectual disability (n = 3), and skeletal abnormalities (n = 1) (Figure 2).
Figure 2
Clinical images showing dysmorphic features of all patients. A−C. Noonan syndrome (patients 1–3) – triangular facies, hypertelorism and epicanthal folds. D. Silver–Russell syndrome (patient 4) – triangular facies, flat facial profile, and relative macrocephaly.E−G. Prader–Willi syndrome (patients 5–7 ) – obese phenotype, narrow bifrontal diameter, almond shaped eyes and lipomastia/gynecomastia. H. 3M syndrome (patient 8) – large head, long philtrum and short broad neck. I. KBG syndrome (patient 9)– triangular facies, bushy eyebrows, macrodontia, bulbous nose. J. BPES syndrome (patient 10) – blepharophimosis, ptosis, epicanthus inversus and hypertelorism. K. 18-P deletion syndrome (patient 11) – low nasal bridge, large protruding ears, dental anomalies in 18-P deletion syndrome. L. bone-marrow failure syndrome 3 (patient 12) – broad nasal bridge, long philtrum, frontal bossing. M. desmosterolosis (patient 13) – microcephaly, tall forehead, micrognathia, hearing loss with hearing aid. N. Poirier–Bienvenu neurodevelopmental syndrome (patient 14) – broad forehead with depressed nasal bridge. O. Costello syndrome (patient 15) – epicanthal folds, thick eyebrows, low-set ears. P. Warsaw breakage syndrome (patient 16) – microcephaly, low set ears, beaked nose
All children had proportionate SS. The median peak GH level after provocative GH testing was 4.75 ng/ml (range 0.20–27.82 ng/ml). Among the 16 children evaluated, 12 had peak GH levels < 7 ng/ml; of these, 7 had low IGF-1 and 5 had normal IGF-1 levels. One child with GH between 7 and 10 ng/ml had normal IGF-1, while among those with GH > 10 ng/ml, one had low, one had normal, and one had elevated IGF-1 levels. The recognizable syndromes noted in our study cohort were Noonan syndrome, Prader–Willi syndrome, SRS, and blepharophimosis-ptosis-epicanthus inversus syndrome (BPES). Central hypogonadism was noted in all PWS cases. Hypergonadotropic hypogonadism was seen in 3M and BPES. Panhypopituitarism was observed in 18p deletion syndrome. The auxological data, clinical characteristics, and details of rhGH therapy for all children are summarized in Tables I and II. PWS and SRS were diagnosed by multiplex ligation-dependent probe amplification (MLPA) testing, and the remainder were diagnosed by NGS. The molecular genetic analysis of all 16 study cohorts is detailed in Table III.
Table I
Baseline clinical and biochemical characteristics of children with syndromic short stature (SSS)
[i] ASD – atrial septal defect; GH – growth hormone; IGF-1 – insulin-like growth factor; IUGR – intrauterine growth retardation; KBG – KBG syndrome; MRI – magnetic resonance imaging;
SDS – standard deviation score; SGA – small for gestational age; SNHL – sensorineural hearing loss; 3M – Miller–McKusick–Malvaux
Table II
Growth hormone therapy details and first-year treatment outcomes
Table III
Molecular analysis of syndromic short stature cases
[i] AD – autosomal dominant; ANKRD11 – ankyrin repeat domain-containing protein 11; AR – autosomal recessive; CUL7 – cullin 7; CSKN2B – casein kinase 2 beta polypeptide; DDX11 – DEAD/
H-box helicase 11; DHCR24 – 24-dehydrocholesterol reductase; DNAJC21 – DnaJ heat shock protein family (Hsp40) member C21; Het – heterozygous; Hom – homozygous; HRAS – Harvey rat sarcoma viral oncogene homolog; MS-MLPA – methylation-specific multiplex ligation-dependent probe amplification; PM – pathogenic moderate; PP – pathogenic supporting; PS – pathogenic strong; PTPN11 – protein tyrosine phosphatase, non-receptor type 11; PVS – pathogenic very strong; VUS – variant of uncertain significance; WES – whole exome sequencing
The mean height velocity before rhGH therapy was 3.99 ±0.54 cm/year. The mean height velocity after 1 year of rhGH therapy in the overall cohort was 7.47 ±1.69 cm/year (p = 0.01), and the median height SDS improved to –3.62 (IQR: –4.74 to –2.73). The 18p deletion case exhibited normal IGF-1 (192.2 ng/ml) and low peak GH (0.20 ng/ml), achieving the maximum height gain (13 cm) among the whole cohort. In contrast, the 3M syndrome child showed elevated IGF-1 (502.1 ng/ml) and peak GH (19.2 ng/ml) despite a lower height velocity of 6 cm/year, suggesting partial GH/IGF-1 resistance. The median [IQR] change in height SDS (Δ height SDS) after one year of rhGH therapy was +0.57 [0.47–0.71] (95% CI 0.33–0.89, p = 0.003). None of the children had GH-related adverse effects. All children received supportive multidisciplinary management and advice from various specialists, tailored to their co-morbidities and coexisting features. Reproductive counseling and the option of prenatal tests could be given in some of the cases where an underlying variant/mutation was identified.
Discussion
SS is an important clinical feature of most genetic syndromes. The clinical suspicion of SSS arises when there are dysmorphic facies with severe SS, parental consanguinity, and/or SGA and/or intrauterine growth restriction (IUGR) at birth, in the background of a normal GH provocative test. The appropriate diagnosis of genetic etiologies in SSS will help elucidate the molecular genetic basis, guide recognition of associated clinical conditions, delineate etiologies, and support genetic counseling [9, 10]. DNA repair disorders such as Bloom’s syndrome, are associated with oncogenic risk, especially hematological malignancies, where rhGH is contraindicated due to potential tumor promotion. This article aims to explore the etiological diagnosis of SSS, focusing on its clinical and genetic heterogeneity and discussing differential responses to rhGH therapy based on precise molecular diagnosis.
The prevalence of SS in India ranges from 2.8% to 3.7% [11]. Given the greater number of consanguineous marriages and regional diverse ethnicity in India, with a variable genetic background, syndromic etiologies of SS are more likely to be prevalent. Therefore, any case labelled as ISS should undergo thorough evaluation to rule out syndromic features, including comprehensive genetic testing. The overall prevalence of SSS is expected to be around 8–10%, with even higher rates observed in studies of pathological short stature. Case series and studies from Northern India have identified a broad spectrum of underlying syndromes, ranging from common etiologies such as Turner’s, Noonan’s, and SRSs to rare entities like skeletal dysplasia and lysosomal storage disorders [12–14]. In the present study, Turner’s syndrome was excluded due to its well-established phenotype and karyotype correlation worldwide. Similarly, lysosomal storage disorders and all skeletal dysplasias were excluded due to their heterogeneous clinical characteristics and distinct etiological profiles.
Currently, there is no universally accepted classification of SSS. A few authors have proposed dividing the SSS into two main groups based on the presence or absence of IUGR/SGA at birth [15]. The presence of SGA serves as a significant clinical indicator and may suggest an underlying syndromic form of SS. SGA has been documented in a wide range of genetic syndromes associated with SS. In children born with SGA, the frequency of underlying genetic mutations is higher than in ISS. The diagnostic yield of identifiable genetic abnormalities in SGA children varies across studies, ranging from approximately 15% to 25% [16, 17]. In addition, SGA without catch-up growth itself is an approved indication for rhGH therapy. In the present cohort, 50% of SSS children had SGA/IUGR. Henceforth, the birth history is an essential component of SSS evaluation. Moreover, the greater the severity of SS in SGA, the higher the likelihood of detecting a specific genetic defect. Therefore, SS with SGA is an appropriate indication for genetic testing in the context of SSS, with or without a characteristic phenotype.
Genetic evaluation of SSS employs various modalities, including conventional karyotype, MLPA, chromosomal microarray analysis (CMA), single-gene sequencing, NGS, and clinical exome sequencing. Each technique has its own advantages and limitations for diagnosing the genetic etiology of SSS. A simplified approach to suspect SSS involves classifying them into proportionate and disproportionate short-stature patterns (Figure 1). In cases of disproportionate SS, skeletal dysplasia is usually suspected; a skeletal survey is recommended, followed by targeted genetic testing guided by the radiographic diagnosis. In cases of proportionate SS, if a recognizable syndromic phenotype is evident, specific genetic testing based on clinical suspicion is advised. If the phenotype is unrecognized, CMA or comparative genomic hybridization is the first-line investigation; if these studies are normal, further evaluation with NGS and methylation-specific testing is pursued according to the clinical context [18, 19]. The diagnostic yield of whole-exome sequencing (WES) in SSS has been reported to range between 25% and 50% [5]. WES is often phenotype-driven (clinical–exome approach) and can be expanded to a complete exome if needed. Owing to financial constraints, CMA was not performed before WES in the present series. WES with the ExomeDepth algorithm was used to detect single-nucleotide variants (SNVs) and copy number variants (CNVs) simultaneously. CNV calls were supported by the CNV ratio and Bayes factor. Orthogonal confirmation was not feasible due to financial constraints. Given the clinical suspicion of a monogenic disorder in most cases, WES was prioritized as a cost-effective diagnostic approach. Moreover, with advances in bioinformatic algorithms, WES is increasingly replacing CMA as the initial genetic testing approach for evaluating unexplained SS, given its comparable sensitivity for detecting CNVs [20]. In recent years, near-total concordance (98.9%) between WES and CMA has been reported; additionally, WES has the added diagnostic power to identify SNVs, CNVs, uniparental disomy (UPD), and trisomy [21]. Overall, WES offers a significantly higher diagnostic yield (~27%) than CMA (~13.6%). In the current scenario, CMA may be considered if WES is inconclusive or if significant CNVs are suspected. Hence, appropriate clinical phenotyping is mandatory to choose appropriate genetic testing.
Although GHD is not a documented pathophysiological mechanism in most cases, a few of the SSS had GH/IGF-1 axis involvement. The Food and Drug Administration has approved GH therapy for various syndromic etiologies such as Turner syndrome, Noonan syndrome, SRS, and SHOX haploinsufficiency. Although molecular pathways and cellular mechanisms, including those involved in growth signaling, have been elucidated for many syndromes, the options for targeted therapies remain limited. GH therapy remains one of the widely available growth-promoting treatment options to date; hence, a trial of rhGH therapy may be warranted. The largest cohort study with extended longitudinal follow-up on the safety and adverse effects of rhGH in children, the KIGS cohort study, supports its use across a range of GHD and non-GHD conditions [22]. Overall, rhGH use in SSS should be individualized, guided by clinical phenotype, underlying pathophysiology, and the feasibility of long-term surveillance. The use of rhGH therapy in various syndromic etiologies is summarized in Figure 3.
Figure 3
Syndromic short stature and prioritization of recombinant growth hormone therapy based on current evidence and safety Considerations
CMA – chromosomal microarray analysis; FDA – Food and Drug Administration; GH – growth hormone; IGF-1 – insulin-like growth factor 1;
MRI – magnetic resonance imaging; MS-MLPA – methylation-specific multiplex ligation-dependent probe amplification; rhGH – recombinant human growth hormone; SHOX – short stature homeobox gene; WES – whole exome sequencing

In the present study, children with Noonan syndrome (Figure 2A,1B,1C) demonstrated a clinically relevant improvement after 1 year of rhGH therapy with a first-year growth velocity of around 7.4–8 cm/year (children 1–3). A pathogenic PTPN11 mutation was noted in all three reported cases. Mutations in several genes, such as PTPN11 (50 %), SOS1 (10–15%), RAF1 (5–10%), KRAS, and NRAS, are reported in Noonan syndrome. These genetic mutations are associated with alterations in the RAS-mitogen-activated protein kinase signaling pathway, disrupting normal cellular processes involved in growth. Despite the suboptimal overall growth response in Noonan syndrome with PTPN11 mutations due to intrinsic GH resistance, as reported in the literature, our series showed a reasonable response [23]. As this condition falls under RASopathies, a predisposition to tumors involving different organ systems is a remote possibility; therefore, routine tumor surveillance is recommended. Both hematological and solid-organ tumors have been reported with Noonan syndrome. Clinical vigilance remains the mainstay of tumor surveillance; any new findings, such as unexplained pallor, bruising, persistent fever, lymphadenopathy, or abdominal distention, should prompt evaluation and referral to the hematology/oncology team. Additionally, ultrasound of the abdomen, CBC, and peripheral smear may be performed every 6 months as part of tumor surveillance. In Noonan syndrome, rhGH therapy should be undertaken with caution in the presence of hypertrophic cardiomyopathy, although it does not represent an absolute contraindication [24].
SRS (Figure 2D) is a prototypical form of SSS with IUGR (child 4). GH therapy will be beneficial for height gain in SRS, with greater improvements in younger children who initiate therapy at a younger age. Our child demonstrated a favorable response. An increase in height velocity of ≥ 3 cm per year is an expected growth response to rhGH in SRS [25].
Prader–Willi syndrome (children 5–7) is a unique SSS in which SS is associated with obesity, hyperphagia, hypotonia, hypogonadism, features such as micropenis with or without cryptorchidism, and mental disability [26]. All of our PWS children (Figure 2E, 1F, 1G) had demonstrated a clinically meaningful response to rhGH therapy, with height gain of 8–9 cm in the first year, with no safety concerns.
3M syndrome (child 8) is an autosomal recessive disorder widely reported worldwide, characterized by severe postnatal and prenatal growth retardation and dysmorphic features. Mutations in the CUL7 gene (77%) are the primary cause and were identified in our patient, with OBSL1 mutations (16%) accounting for a smaller proportion. Both genes are key components of the ubiquitin-proteasome system and cytoskeletal architecture and are involved in chondrocyte function. The SS in 3M syndrome is mainly due to partial GH resistance rather than GHD. Clinical studies indicate that GH therapy is only moderately effective, while early initiation and prolonged treatment may offer modest benefits. A notable case reported by Deeb et al. [27] showed that high-dose GH (0.045 mg/kg/day) improved height SDS from –5.3 to –3.9 over 3 years in a child with a CUL7 mutation. The child in our cohort (Figure 2H) presented with a height SDS of –5.5. During the first year of GH therapy, the height velocity was 6 cm, the lowest among all SSS in the present cohort. The child had high GH and IGF-1 levels both at baseline and during rhGH therapy. Despite receiving a higher rhGH dose (0.40 mg/kg/week), growth was suboptimal. This suggests partial GH or IGF-1 resistance, possibly due to defects in downstream signaling pathways. Similar findings have been reported in 3M and have even been documented as a syndrome resembling GH insensitivity phenotypes [28]. The other coexistent endocrine manifestation of 3M syndrome was hypergonadotropic hypogonadism.
KBG syndrome (child 9) is a rare genetic disorder, with approximately 100 cases reported globally, caused by mutations in the ANKRD11 gene, with characteristic craniofacial features. This gene encodes a nuclear protein, and it is essential for the development of multiple systems. Several reports have documented the use of GH therapy in KBG syndrome, with variable outcomes. According to the available literature, the mean height SDS in children with KBG syndrome and SS improved significantly from –2.72 ±0.44 to –1.95 ±0.57 after the first year of GH therapy (p = 0.001), with no reported adverse effects [29]. The child in our cohort (Figure 2I) harbored a pathogenic mutation within a known ANKRD11 mutational hotspot, presenting at 6 years of age with severe SS (height SDS –5.5) and a clonidine-stimulated GH peak of 5.5 ng/ml. After one year of GH therapy, the height SDS improved from –5.2 to –4.7 with height gain of 7.2 cm.
Child 10 with BPES, a rare genetic disorder caused by mutations in the FOXL2 gene, presented at 13.2 years with severe SS (height SDS: –4.2) and dysmorphic facial features of BPES. GH stimulation testing revealed a subnormal peak GH level of 2.45 ng/ml, confirming GHD. The second common endocrine manifestation noted in BPES was primary ovarian failure. Therefore, in a child presenting with SS and hypergonadotropic hypogonadism accompanied by dysmorphic features, after ruling out Turner syndrome, SSS such as BPES or 3M syndrome may be suspected. Following initiation of GH therapy, her first-year height velocity was 7 cm, with an improvement in height SDS from –4.2 to approximately –3.4. Hormone replacement therapy for pubertal induction was initiated concurrently. BPES is classified into two types: Type I, which is associated with primary ovarian insufficiency, and Type II, which presents without gonadal involvement. IGHD has also been reported to be associated with BPES, and a beneficial response to rhGH therapy has been documented [30].
18p deletion syndrome is a rare chromosomal disorder characterized by deletion of the short arm of chromosome 18, noted in case 11 of our SSS cohort, with a remarkable growth response to rhGH. Her height velocity (HV) increased to 13 cm in the first year of treatment (+1.4 SDS gain). Additionally, she had panhypopituitarism; steroid and levothyroxine replacement therapies were initiated at baseline. Pituitary hypoplasia was noted in the MRI pituitary. This case underscores the importance of considering GH with panhypopituitarism as a contributing factor to SS in children, even when a genetic syndrome is present and growth retardation cannot be attributed to the syndromic background alone. According to the literature, GH therapy can significantly improve linear growth in patients with 18p deletion syndrome [31]. Early recognition and treatment of GHD can optimize growth outcomes in these patients. Similarly, pituitary stalk interruption syndrome is a well-recognized radiological and clinical entity that often serves as a clue to other syndromic etiologies of CPHD, presenting with midline anomalies and varying degrees of pituitary hormonal deficiencies, and rhGH therapy is usually associated with a robust therapeutic response [32].
One of our children in the present cohort, diagnosed with bone marrow failure syndrome-3 (child 12), a rare disorder caused by mutations in the DNAJC21 gene, presented with severe SS and cytopenia. GH therapy led to significant height gain. Another case involved a patient with desmosterolosis (child 13), an infrequent autosomal recessive disorder affecting cholesterol biosynthesis, characterized by multiple congenital anomalies, developmental delay, and growth failure. The efficacy and safety of rhGH therapy in this syndrome are yet to be elucidated, though a child in our cohort showed a reasonable response compared to pretreatment HV. Furthermore, a child with Poirier–Bienvenu neurodevelopmental syndrome (child 14), a disorder caused by mutations in the CSNK2B gene and marked by intellectual disability and epilepsy, demonstrated an optimal growth response to GH treatment. Biochemical evidence of GHD was also noted. These cases underscore the importance of considering GH therapy in patients with rare genetic syndromes, such as Shwachman–Diamond syndrome [33], where GHD can coexist with other systemic features. Similarly, patients with CSNK2B deficiency may benefit from GH therapy [34]. Costello syndrome (child 15), a RASopathy similar to Noonan syndrome, is caused by an activating mutation of the proto-oncogene HRAS. The usual presentation is severe growth retardation with multi-system abnormalities, though the phenotypic spectrum is broad. Warsaw breakage syndrome (child 16) is characterized by variants in the DDX11 gene, which plays a crucial role in proper cell division through chromatid cohesion. rhGH therapy has been initiated under close tumor surveillance in patients with Costello, Warsaw breakage, and bone marrow failure syndrome, as supported by current literature, given the potential oncogenic risk associated with these underlying genetic defects. Abdominal ultrasound, CBC, peripheral smear study, and determination of serum lactate dehydrogenase (LDH) and alpha-fetoprotein (AFP) levels were performed every 6 months. A team of hematologists, oncologists, and endocrinologists regularly followed all of these patients.
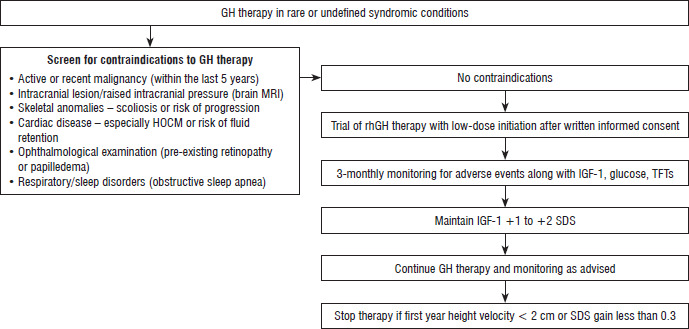
The median height SDS gain of +0.57 [0.47–0.71] (95% CI: 0.33–0.89, p = 0.003) over 1 year is considered clinically meaningful, given the low pretreatment height velocity and the heterogeneous syndromic background of the cohort. This suggests that a trial of GH therapy may be considered in selected syndromic cases, provided potential complications – such as scoliosis or other active malignancies that could be exacerbated by rhGH treatment – are absent. Figure 4 illustrates the clinical decision framework preceding GH initiation in cases of syndromic or atypical SS with an undefined underlying diagnosis.
Figure 4
Clinical decision framework preceding GH initiation in cases of syndromic or atypical short stature where the underlying diagnosis remains undefined
GH – growth hormone; HOCM – hypertrophic obstructive cardiomyopathy; IGF-1 – insulin-like growth factor 1; MRI – magnetic resonance imaging; rhGH – recombinant human growth hormone; SDS – standard deviation score; TFTs – thyroid function tests
Interestingly, in our cohort, 12 children demonstrated peak GH levels below 7 ng/ml, of whom 5 exhibited normal IGF-1 concentrations. Conversely, among those with GH peaks exceeding 10 ng/ml, IGF-1 responses were variable – one normal, one low, and one elevated. Given that IGF-1 secretion is influenced by multiple confounding factors such as nutritional status, sampling timing, and physiological state, particularly in syndromic contexts, this discordance may suggest a spectrum of perturbations. These may range from partial GH–IGF-1 axis involvement or partial resistance to defects downstream of GH signaling, including intracellular pathways regulating chondrocyte proliferation and differentiation. The heterogeneity observed underscores the complex biology underlying growth failure in SSS. In the absence of other established molecular or growth-promoting therapies, a carefully monitored therapeutic trial of rhGH may be justifiable.
The strength of this study is that it boasts a robust cohort of children with diverse SSS etiology, and all of them underwent comprehensive molecular genetic analysis. All patients received rhGH therapy and were meticulously followed up to assess the clinical response. The clinical predictors of GH therapy response with molecular genetic diagnosis were analyzed across the different etiological spectrum of SSS.
Limitations
Limitations are that trio-WES and segregation analysis did not validate variants of uncertain significance (VUS). However, VUS was considered in this study only when the relevant phenotype was matched, and no other variants pertinent to the patient’s phenotype were detected. Moreover, in silico predictions of the detected VUSs were damaging in all cases. In the absence of trio WES, mild variant misclassification is a possibility; however, clinical decisions, including GH therapy initiation, were primarily based on auxological and phenotypic criteria, minimizing the potential impact of this technical limitation on outcome interpretation. Another limitation is the use of a fixed dose of rhGH therapy, as optimal dosing for each syndrome remains unclear. Moreover, each syndrome is molecularly heterogeneous, with distinct genetic defects and pathophysiological mechanisms contributing to growth failure. As such, grouping them may limit the coherence and generalizability of the findings. The absence of an untreated control group – or an appropriately matched historical control – makes it difficult to attribute the observed improvement in growth exclusively to rhGH therapy. To gain a more thorough understanding of the long-term efficacy and potential risks associated with rhGH therapy in this population, extended follow-up and multicenter studies are essential.
Conclusions
SGA and IUGR with dysmorphic facies may serve as early clinical indicators of syndromic etiology of SS, prompting molecular genetic analysis. WES is vital for diagnosing subtle or unrecognized syndromes misclassified as ISS or SGA-related SS. RhGH therapy improves the height velocity in a diverse spectrum of SSS. Increased use of GH therapy across various SS syndromes adds to the global knowledge of GH therapy in syndromic etiologies. However, in cases associated with tumorigenesis, great caution should be exercised. Multicenter studies are needed to define the long-term role of GH therapy in these genetic conditions.









