

Introduction
Autoimmune lymphoproliferative syndrome (ALPS) is a chronic non-malignant lymphoproliferative disorder caused by mutations in the genes involved in programmed cell death [1-4]. In the majority of patients, ALPS is due to inherited mutations in genes encoding proteins of the Fas pathway, which mediates programmed cell death or apoptosis. Several genetic defects are associated with ALPS. ALPS-FAS and ALPS-s FAS are caused by germline and somatic mutations, respectively, in the FAS gene [5-9]. In rare cases, ALPS is caused by mutations in the genes encoding the Fas ligand (ALPS-FASLG) or caspase 10 (ALPS-CASP10). It is inherited as an autosomal dominant pattern with variable penetrance. As a very rare disease, ALPS was first characterized in the early 1990s. The incidence and prevalence of ALPS are unknown. Estimated cases of ALPS worldwide exceed several hundred, but that number has not reliably been confirmed. Cases of ALPS probably remain undiagnosed due to variable phenotypic expression and a constellation of symptoms that overlap with many other conditions, particularly Evans syndrome and other lymphoproliferative disorders [10, 11]. Its clinical manifestations include: unexplained lymphadenopathy, hepatosplenomegaly, autoimmune cytopenia such as thrombocytopenia, neutropenia, and anemia due to excessive production of antibodies by lymphocytes, elevated number of double-negative T (DNT) cells, and increased risk of lymphoma [12-15]. The diagnostic approach to these patients tends to be broad and often includes imaging studies as well as invasive diagnostic procedures, such as biopsy of lymph nodes and/or other tissues. Molecular characterization of ALPS has resulted in revised diagnostic criteria, aiding in the improvement of ALPS diagnosis and treatment. The revised 2010 international consensus diagnostic guidelines incorporated both phenotypic manifestations and genotype [16]. This revision divides diagnostic criteria for ALPS into 2 required and 6 accessory criteria. Required criteria include the presence of lymphadenopathy and/or splenomegaly, and elevated TCRa/b+ cells. Accessory criteria are subdivided into primary, which include an abnormal lymphocyte apoptosis assay and the presence of pathogenic mutations in genes of the FAS pathway; and secondary, which include the presence of elevated circulating biomarkers, characteristic histopathology, and the combined presence of autoimmune cytopenias, polyclonal hypergammaglobulinemia, and family history compatible with ALPS. For ALPS diagnosis a patient has to meet both required criteria and one of the primary accessory criteria. A probable ALPS diagnosis can be entertained by the presence of the required criteria and any one of the secondary accessory criteria. The modifications in the diagnostic criteria and classification system should facilitate diagnosis and allow easy inclusion of newly discovered genetic defects resulting in classical ALPS [16-18]. ALPS is reported in various racial and ethnic backgrounds. Male predominance was previously suggested. This gender inequality was confirmed in both the French ALPS cohort and the National Institutes of Health (NIH) ALPS cohort [18, 19]. In this paper we present the first report of a Macedonian family with ALPS, caused by a novel heterozygous mutation in the FAS gene identified through reverse phenotyping.
Case 1
A 14-month-old Macedonian girl was referred to our clinic due to fever, pallor, anemia, thrombocytopenia and splenomegaly. The child was an offspring of non-consanguineous parents, born at 40 weeks of gestation with birth weight 3180 g and body length 50 cm. Past medical history revealed anemia at the age of 2 months found on a routine check-up, gastrointestinal infection two months before the hospitalization and subsequently measles infection, after which she developed severe anemia. Family history provided data for splenomegaly of unknown cause and subsequent splenectomy in the patient’s mother at the age of 14. Hereditary spherocytosis was an initial diagnostic consideration. Physical examination at the admission revealed pallor, skin and mucous hemorrhages, significant splenomegaly of 128 cm (380 cm3), and lower limb edema. Analysis of the complete blood count (CBC) showed: hemoglobin (Hb) 7.1 g/dl, red blood cells (RBC) 2.95 × 1012/l, white blood cells (WBC) 10.2 × 109/l, platelets (PLT) 7 × 109/l, and reticulocyte count at 3%. Due to the family history of splenectomy in the patient’s mother and suspicion of spherocytosis, genetic analysis was performed.
Case 2
The second patient is the second child in the same family, female, born at term with birth weight 3090 g and body length 50 cm. The patient was hospitalized with severe anemia at the age of 1.5 months, with Hb 4.6 g/dl, RBC 1.75 × 1012/l, WBC 10.8 × 109/l, PLT 129 × 109/l. Physical examination at admission revealed skin pallor, without organomegaly or other symptoms of the disease. Due to ALPS in the family, genetic analysis was done and ALPS diagnosis was confirmed.
Method for diagnosis
Genetic analysis of the proband included next generation sequencing (NGS) on a MiSeq personal sequencer using the Illumina TruSight One kit. Direct DNA sequencing of FAS exon 9 was performed for the proband, her parents and grandparents. In addition, complete laboratory analysis was performed, including: CBC, detection of anti-RBC antibody, immunoglobulins (immunoglobulin (Ig)G, IgM, IgA), vitamin B12, double negative T (DNT), total proteins, albumins and triglycerides.
Results
Genetic analysis showed no pathogenic variants in the genes associated with spherocytosis. However, a novel pathogenic variant, c.913dupA, p.Thr305AsnfsTer16 in exon 9 of the FAS gene, was revealed. The same mutation was present in the patient’s mother, but not in her parents (proband’s grandparents). Thus, the pathogenic FAS variant has arisen as a de novo event in the proband’s mother (Fig. 1). Direct sequencing results of exon 9 of the FAS gene are given in Figure 2. Additional laboratory investigations showed increased numbers of CD3+CD4–CD8– T cells (28%), Coombs-positive hemolytic anemia, severe thrombocytopenia, specific biomarkers for ALPS which included hypergammaglobulinemia, elevated level of vitamin B12, hypertriglyceridemia, and hypoalbuminemia. In addition, the proband was found to be a silent carrier of alpha thalassemia (α-3.7) inherited from her father. Clinical findings and laboratory results of the patients are given in Table 1.
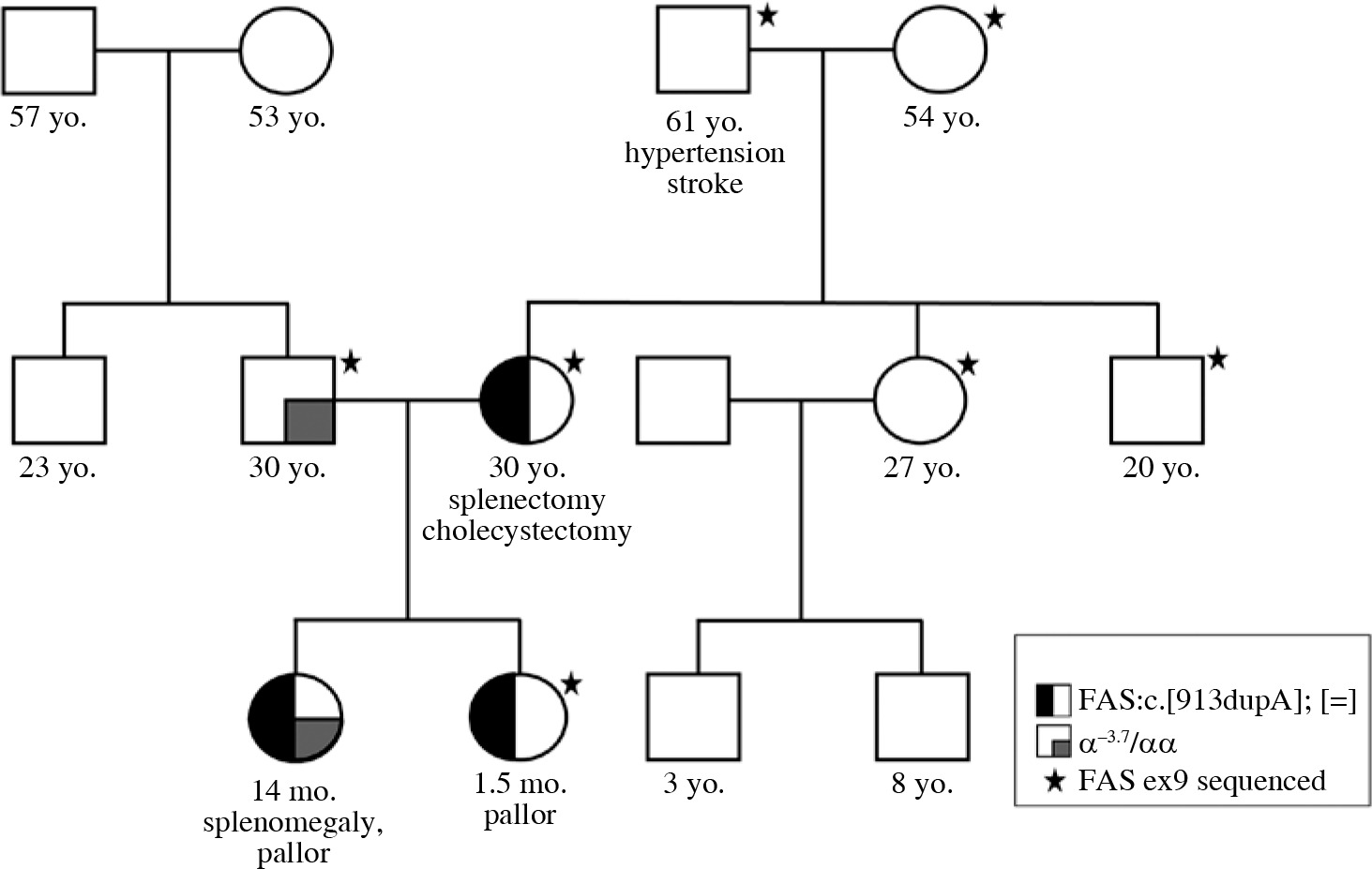
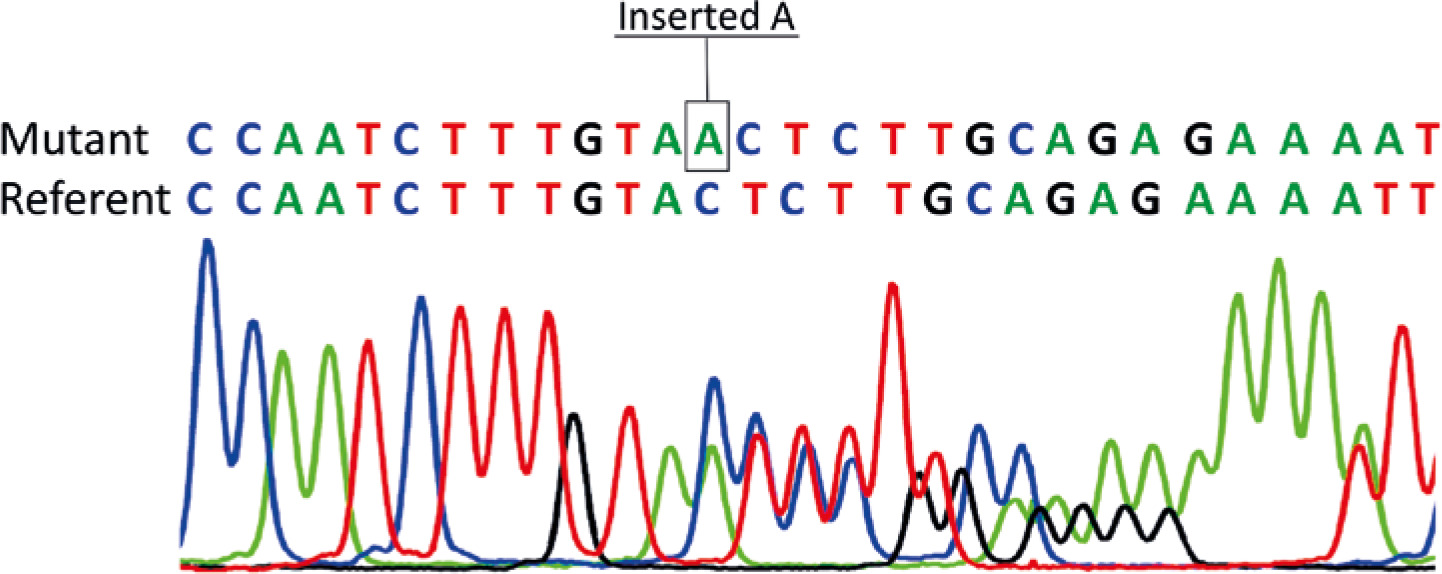
Table 1
Clinical findings and laboratory test results of the patients
Discussion
To the best of our knowledge, this the first case of ALPS reported from our country. The patient was diagnosed to have ALPS on the basis of reverse genotyping. A first-line NGS analysis in our case allowed identification of a novel pathogenic variant in the FAS gene and initiation of an appropriate clinical examination to promptly establish the clinical diagnosis and initiate specific therapy early. Genetic analysis also revealed that the proband (case 1) is a silent carrier of α-thalassemia inherited from her father. The proband fulfilled ALPS diagnostic criteria and based on the revised classification of ALPS she has ALPS-FAS type of the disease [16]. She fulfilled two required criteria (splenomegaly and elevated DNT), one accessory primary criterion (germline mutation in the FAS gene) and several accessory secondary criteria (elevated plasma vitamin B12, autoimmune cytopenia, polyclonal hypergammaglobulinemia and positive family history for splenectomy). In addition, we observed hypertriglyceridemia and clinical signs of nephrotic syndrome in the proband, which are rarely observed in ALPS. Thus, ALPS should be considered in differential diagnosis of children presenting with nephrotic syndrome, splenomegaly and unexplained cytopenia. The second child in the family presented with a severe anemia at an early age. Although other ALPS biomarkers were not present at the time of the first manifestation of anemia in this case (case 2), ALPS was confirmed by genetic testing. Thus, the clinical expression of the disease in this family is variable. Treatment in the first case was performed according to the proposed protocol [20, 21] and remission was achieved. The child is receiving regular maintenance with sirolimus.
ALPS is not an acquired disease; it is present at birth, either as an inherited disorder or because of the occurrence of a somatic mutation in the FAS gene, presumed to occur during embryogenesis. Since ALPS cannot be prevented after the birth of an affected individual, in the case of a family history of ALPS, genetic testing and counselling are important. When the genetic cause of the disease is known, family planning and prenatal diagnosis are available. Although genetic counselling was provided and prenatal diagnosis was proposed to the parents, they decided not to undergo genetic prenatal testing.
The prognosis for ALPS patients remains guarded. The new genetic and biological insights have improved the understanding of ALPS. Greater awareness of ALPS should result in a more directed diagnostic approach and earlier initiation of treatment for this disease with promising treatment efficacy.
Conclusions
A first-line NGS analysis allows identification of a genetic defect and initiation of appropriate clinical examination to promptly establish the clinical diagnosis in patients with rare diseases. Reverse phenotyping in our case provided a prompt and accurate diagnosis and early initiation of specific therapy.