Introduction
Exercise training is effective in reducing cardiovascular disease risk factors [1, 2], and it is also an important strategy for the prevention and treatment of cardiovascular diseases [3, 4]. Heart failure (HF) is a complex clinical syndrome representing the end stage of a number of different cardiac diseases. One of the major clinical manifestations of HF is exercise intolerance, which is closely related to poor prognosis in HF patients [5–7]. In HF, exercise training improves patient outcome by increasing exercise tolerance [8, 9] and reducing dyspnea and fatigue [10]. However, its mechanism in HF has not been elucidated.
Brain-derived neurotrophic factor (BDNF), a member of the neurotrophic factor family, has been demonstrated to regulate various neuronal functions including neuroregeneration, neuroprotection, synapse development and plasticity [11]. BDNF is essential for heart development because its genetic deletion leads to the thinning of cardiac chambers, microvascular leakage and, ultimately, early death in mice [12]. In adult mammals, BDNF modulates autonomic transmission to the heart [13] and exerts prominent angiogenic effects [14]. Recent evidence also points to the presence of its receptor, tyrosine receptor kinase B (TrkB), in the myocardium [15, 16]. Serum BDNF levels are decreased and correlated with exercise capacity in HF patients, and have been shown to be increased by exercise training [17, 18]. However, the role of myocardial BDNF/TrkB signaling in cardiac physiology and myocardial response to exercise training, is largely unknown.
Considering that BDNF through AMPK/PGC-1α triggers mitochondrial function of skeletal muscle and exercise training increases BDNF levels in HF patients [19], we hypothesized that activation of the BDNF/TrkB and AMPK/PGC-1α pathway would be involved in exercise training preventing HF induced muscle dysfunction in mice.
Material and methods
Ethics statement
Animal studies were approved by the Ethical Committee of Ethics in Animal Research of the Xin Hua hospital and were conducted in accordance with the International Guiding Principles for Biomedical Research Involving Animals as promulgated by the Society for the Study of Reproduction.
Animals
Eight-week-old male C57BL/6J mice weighing 22–25 g were obtained from Shanghai SLAC Laboratory Animal Company. Mice were housed in a 12 : 12 h dark-light cycle and temperature-controlled environment (22°C) with free access to standard laboratory chow and water. Mice were acclimated for 7 days before any experimental procedures.
Cell culture
C2C12 cells (ATCC, Manassas, VA) were maintained in growth medium consisting of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, USA). Cells were cultured in a humidified incubator under an atmosphere of 5% CO2 at 37°C. Differentiation of myoblasts into myotubes was induced by switching to differentiation medium consisting of DMEM supplemented with 2% horse serum (Gibco, USA) when cells reached 90 to 95% confluency. Myotubes were used for experiments following 4 days of differentiation.
siRNA transfection
Control and BDNF mRNA-specific siRNAs were obtained from Genepharm Company (Shanghai). C2C12 cells cultured in growth medium in 24-well plates were transfected with siRNAs (1 µg per well) using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, Calif, USA). Forty-eight hours after transfection, cells were switched to differentiation medium and cultured for 96 h or otherwise indicated.
Experimental design
Forty mice were assigned to four groups: untrained CON (CON-UT, 10), exercise-trained CON (CON-T, 10), untrained HF (HF-UT, 10) and exercise-trained HF (HF-T, 10). Mice were intubated and were under mechanical ventilation with adequate anesthesia by pentobarbital (50 mg/kg, intraperitoneally). Then, myocardial infarction (MI) was created by ligation of the left coronary artery, as described previously [20]. Excluding the suturing of the coronary artery, the same surgical procedure was performed, and the sham-operated mice were used as the control group. Sham-operated and MI mice were randomly assigned to untrained and exercise-trained groups.
Exercise training protocol
At 4 weeks after MI induction, mice were submitted to moderate-intensity exercise training in swimming warmed-water (30–32°C) apparatus for 60 min, 5 days/week for 8 weeks [21]. The training sessions were performed during the dark cycle of the mice. All untrained mice were placed in the swimming apparatus for 5 min three times a week to become accustomed to the exercise protocol and handling.
Graded treadmill exercise test
Exercise capacity, estimated by total distance run, was evaluated using a graded treadmill exercise protocol for mice as previously described [22]. Briefly, after being adapted to treadmill exercises over 1 week (10 min/day), mice were placed on the treadmill lane and allowed to acclimatize for at least 30 min. Treadmill speed started at 6 m/min and increased by 3 m/min every 3 min with no grade until exhaustion. Tests were performed in sham-operated and MI mice by a blinded observer. Total distance run (meters) was recorded.
Cardiovascular measurements
Heart rate (HR) was determined non-invasively using a computerized tail-cuff system (BP 2000 Visitech Systems, Apex, USA) [21]. Mice were acclimatized to the apparatus during daily sessions over 6 days, 1 week before starting the experimental period. Heart rate measurements were obtained serially in sham-operated and MI mice once a week throughout the 8 weeks of the experiment.
Echocardiographic assessment
Cardiac function was assessed by two-dimensional guided M-mode echocardiography in halothane-anesthetized sham-operated and MI mice. Mice were positioned in the supine position with front paws wide open and an ultrasound transmission gel was applied to the precordium. Echocardiography was performed using an Acuson Sequoia model 512 echocardiography machine (Acuson Corporation, Mountain View, CA, USA) equipped with a 14-MHz linear transducer. Ventricular structure and systolic function were measured as we previously described [23]. Parameters included left ventricular end-diastolic and end-systolic dimensions (LVEDD and LVESD), left ventricular ejection fraction (LVEF) and fractional shortening (LVFS). Heart failure was defined based on the in vivo LV dysfunction (LVEF, 40% in this study).
Plasma norepinephrine levels
Plasma norepinephrine was measured by HPLC using ion-pair reverse-phase chromatography coupled with electrochemical detection (0.5 V) as described by Medeiros et al. [24].
Western blot
Soleus muscle samples were harvested and homogenized on ice in RIPA lysis buffer (Beyotime, Jiangsu, China). Lysates were centrifuged at 12,000 × g for 20 min at 4°C. The protein concentrations were measured using a BCA kit (Pierce Chemical, Rockford, IL, USA). Equivalent amounts of protein extracts (50 µg) were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred onto PVDF membranes (Millipore Corp., Billerica, MA, USA). The membranes were blocked for 1 h with 5% non-fat dry milk in Tris-buffered saline/Tween-20 (TBST) buffer, and then incubated with primary antibodies against BDNF, p-TrkB, p-AMPK and PGC1α (1 : 1000 dilution) (Cell Signaling Technology, Inc., Danvers, MA) at 4°C overnight. Blots were washed three times and incubated with corresponding secondary antibodies for 1 h at room temperature. The signals were developed using enhanced chemiluminescence (ECL) reagent (Tiangen Biotech Co., Ltd., Beijing, China) and visualized with Quantity One software. Targeted bands were normalized to cardiac GAPDH.
RT-PCR
Total RNA was extracted from soleus muscles in each group using Trizol reagent (Invitrogen, Carlsbad, CA, USA). RNA purity was estimated by calculating the 260/280 nm absorbance and reverse transcribed into cDNA using Transcript First-strand cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China) in accordance with the manufacturer’s protocol. RT-PCR was performed using a Super Real PreMix Plus (SYBR Green) kit (Tiangen, Beijing, China) with BDNF gene sets or GAPDH primers. The reactions were performed three times using an ABI Prism 7500 Sequence Detection System (ABI, Foster City, CA, USA). All data from each sample were assessed by 2–ΔΔCt analysis.
Statistical analysis
All values are presented as means ± standard error (SE). Data were tested for normal distribution and one-way analysis of variance (ANOVA) was used to compare all variables between groups. Two-way ANOVA with post hoc Tukey testing was used for multiple comparison purposes. A p-value of < 0.05 was considered statistically significant.
Results
Effect of exercise training on exercise tolerance and plasma norepinephrine levels
After 8 weeks of moderate-intensity exercise training in sham-operated and MI mice, MI mice covered a shorter distance compared to sham-operated counterparts. Of note, exercise in MI mice not only suppressed the decrease in exercise tolerance, but also increased it towards exercise-trained sham-operated mice (Figure 1 A).
Figure 1
Distance run (A) and plasma norepinephrine (NE) levels (B) in untrained and exercise-trained sham-operated mice (CON-UT and CON-T, respectively), and untrained and exercise-trained MI mice (HF-UT and HF-T, respectively). Note that exercise training significantly improved distance run in HF mice
Data are presented as means ± SE. *P < 0.05 vs. CON-UT group.
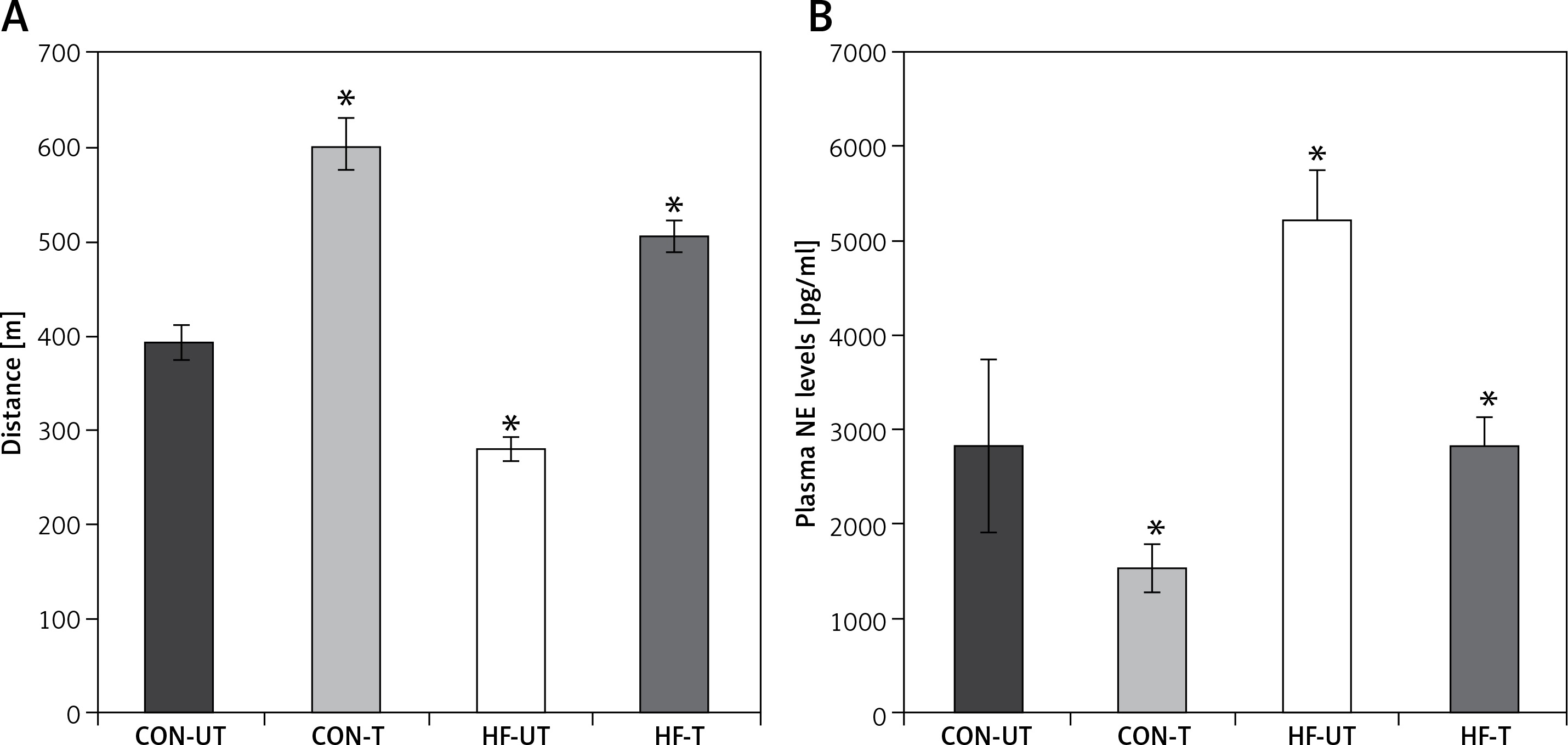
Myocardial infarction mice displayed increased plasma norepinephrine levels compared with sham-operated mice. Exercise training significantly reduced plasma norepinephrine in MI mice toward untrained sham-operated levels (Figure 1 B).
Effect of exercise training on hemodynamic status and LV systolic function
MI mice displayed baseline tachycardia compared with sham-operated mice. Exercise training significantly reduced HR in both sham-operated and MI mice from the third week to the end of the 8-week training period. The reduction of HR in MI was so remarkable that it reached untrained sham-operated levels at the sixth week of training (Figure 2 A).
Figure 2
Heart rate (A), fractional shortening (B) and ejection fraction (C) in untrained and exercise-trained shamoperated mice (CON-UT and CON-T, respectively), and untrained and exercise-trained MI mice (HF-UT and HF-T, respectively). Note that exercise training decreased HR, and increased FS and EF levels in HF mice toward those of untrained CON mice
Data are presented as means ± SE. *P < 0.05 vs. basal levels; #p< 0.05 vs. untrained group.
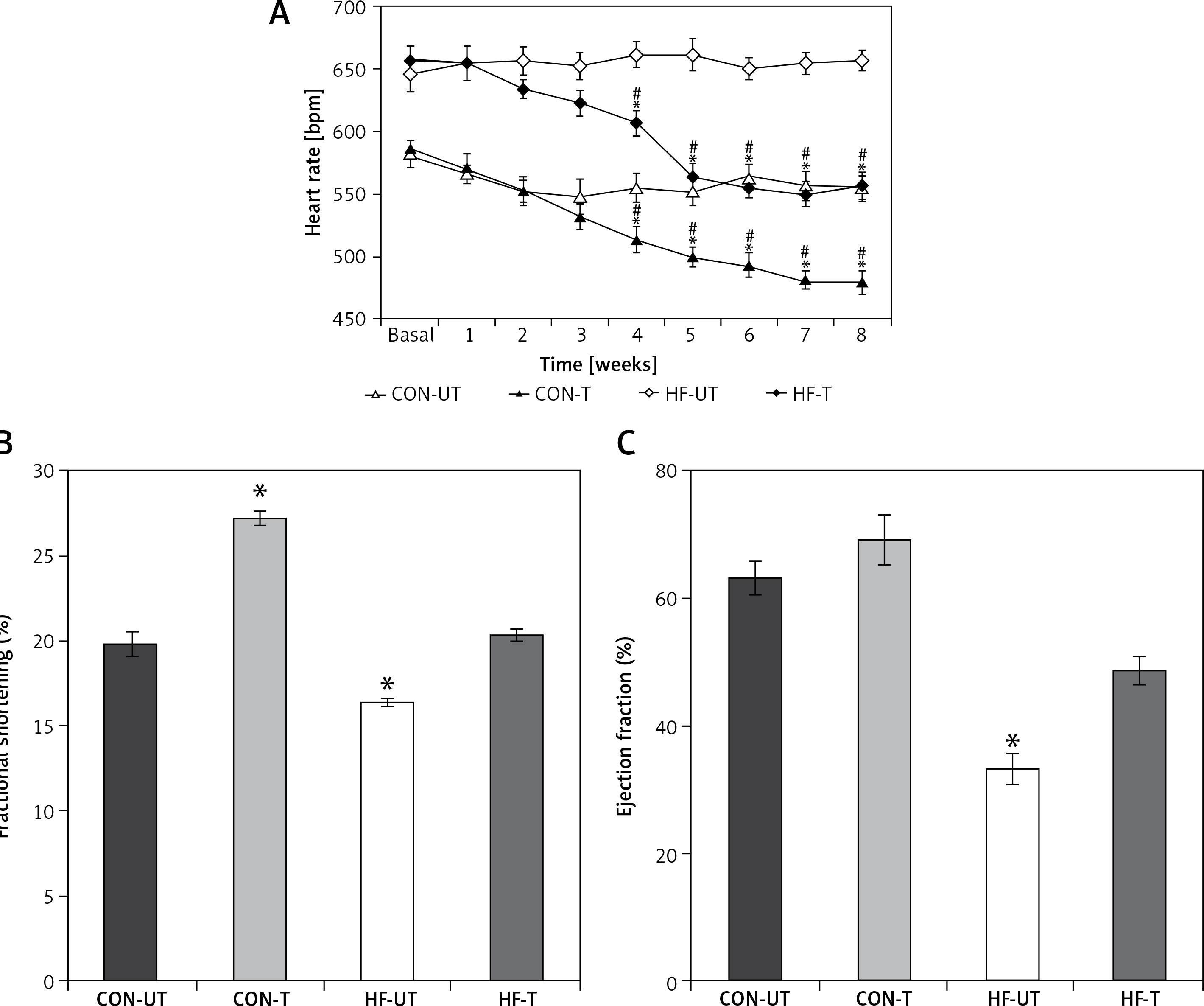
Regarding echocardiographic evaluation, FS and EF were significantly decreased in the MI group compared with the sham-operated group. Eight weeks of exercise training tended to prevent the systolic dysfunction in MI mice (Figures 2 B, C).
Effect of exercise training on BDNF expression
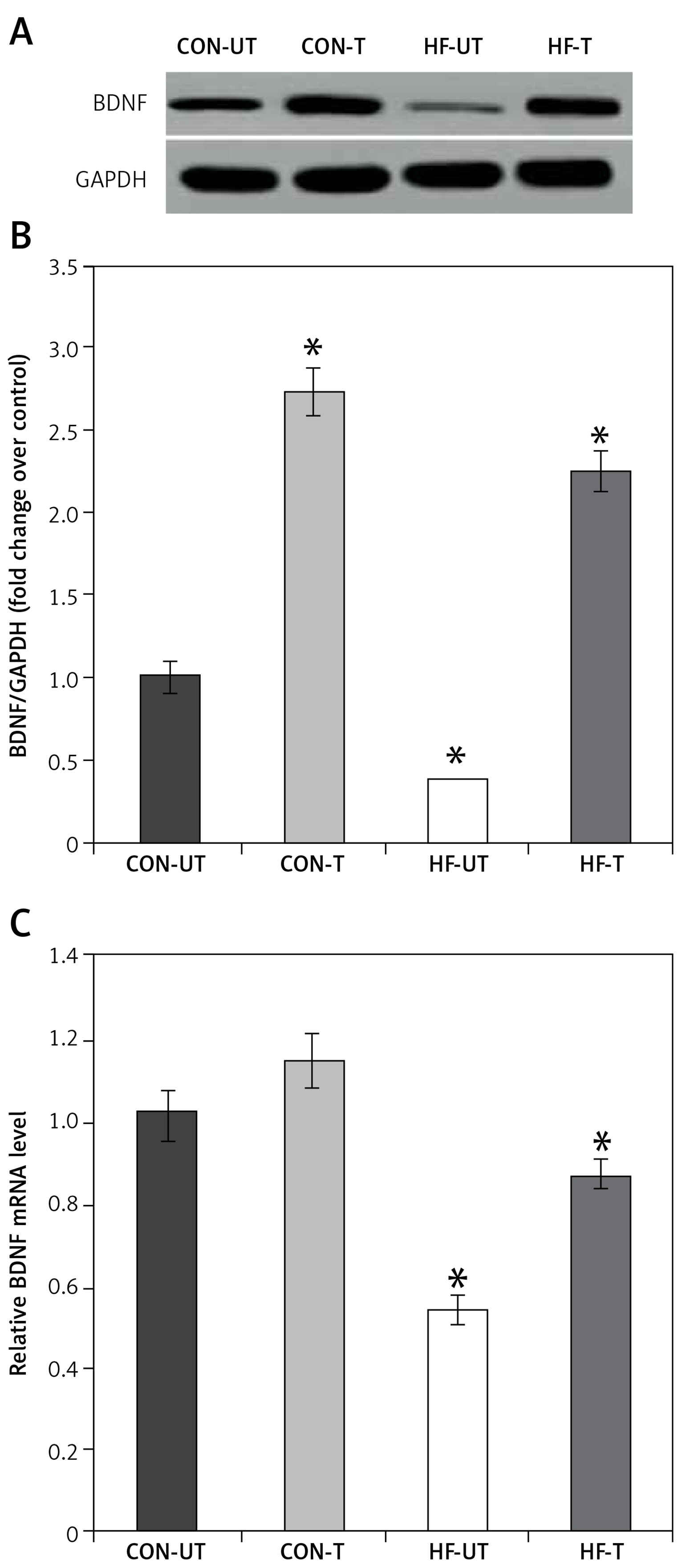
It is well recognized that exercise has a significant impact on BDNF expression, which is involved in neuroplastic processes, contributing to recovery from ischemia [25]. Based on this evidence, we investigated mRNA and protein expression of BDNF in skeletal muscle from sham-operated and MI mice. BDNF mRNA and protein levels were significantly lower in the skeletal muscle from MI compared to sham-operated mice (Figures 3 A–C). However, exercise training prevented the reduction of BDNF content in MI mice (Figures 3 A–C).
Figure 3
BDNF expression in untrained and exercise-trained sham-operated, and MI mice. A – Representative blots of BDNF from untrained (CONUT) and exercise-trained sham-operated (CON-T) and untrained (HF-UT) and trained MI (HF-T) mice. B – Relative protein band density values of BDNF were calculated as the ratio of the protein of interest to that of GAPDH. C – Relative mRNA expression of BDNF
Data are presented as means ± SE. *P < 0.05 vs. CON-UT group.
Effect of exercise training on the components of signaling pathway downstream BDNF
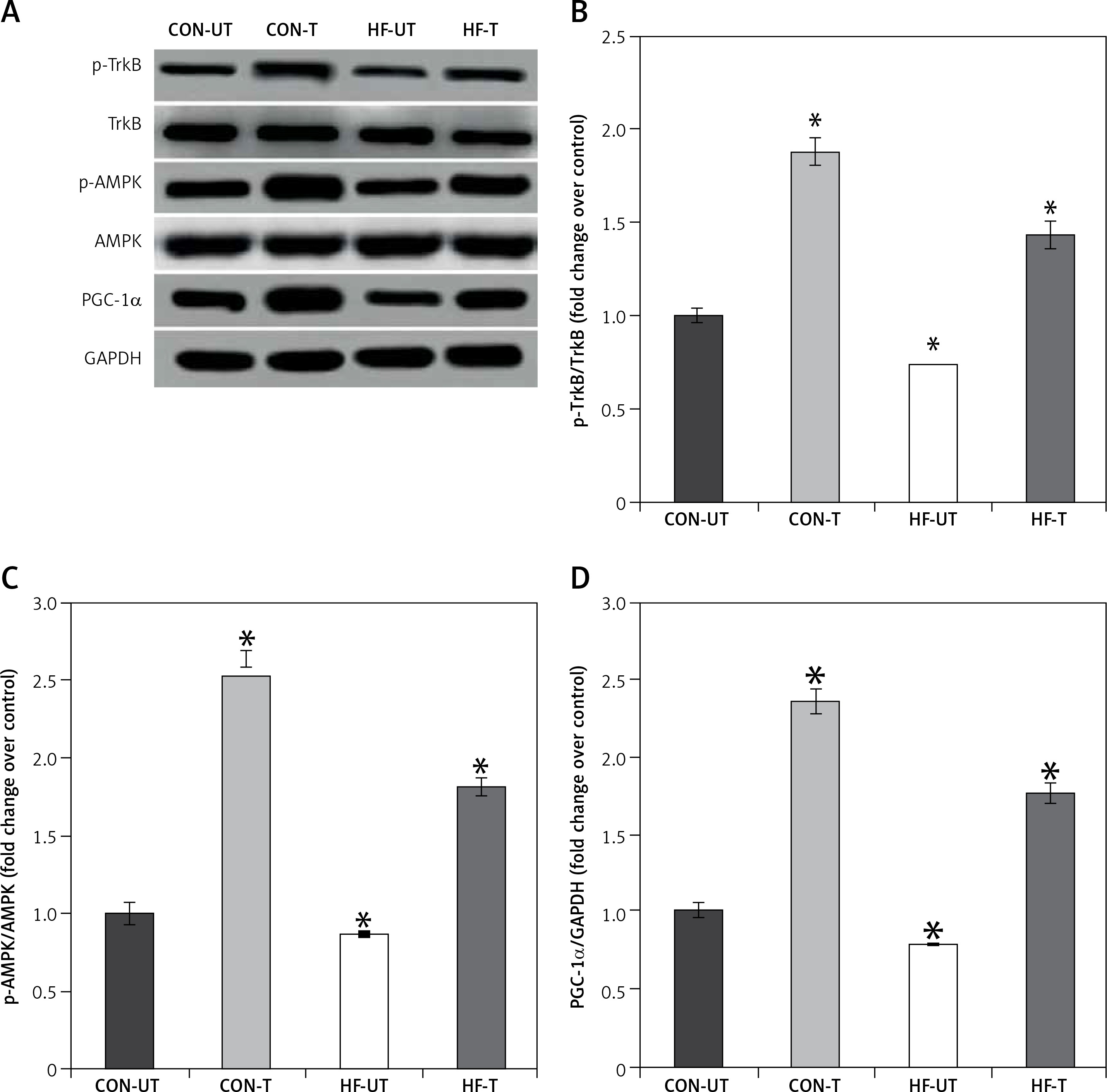
Phosphorylated protein TrkB and AMPK were reduced in the skeletal muscle from MI mice, in association with the decreases in PGC1α levels. Exercise training significantly increased phosphorylated TrkB, AMPK and PGC1α levels in MI mice (Figures 4 A–D).
Figure 4
P-TrkB, p-AMPK and PGC-1α expression in untrained and exercise-trained sham-operated, and MI mice. A – Representative blots of p-TrkB, p-AMPK and PGC-1α from untrained (CON-UT) and exercise-trained sham-operated (CON-T) and untrained (HF-UT) and trained MI (HF-T) mice. B–D – Relative protein band density values of p-TrkB, p-AMPK and PGC-1α were calculated as the ratio of the protein of interest to that of GAPDH
Data are presented as means ± SE. *P < 0.05 vs. CON-UT group.
Effect of BDNF inhibition during myogenic differentiation of C2C12 cells
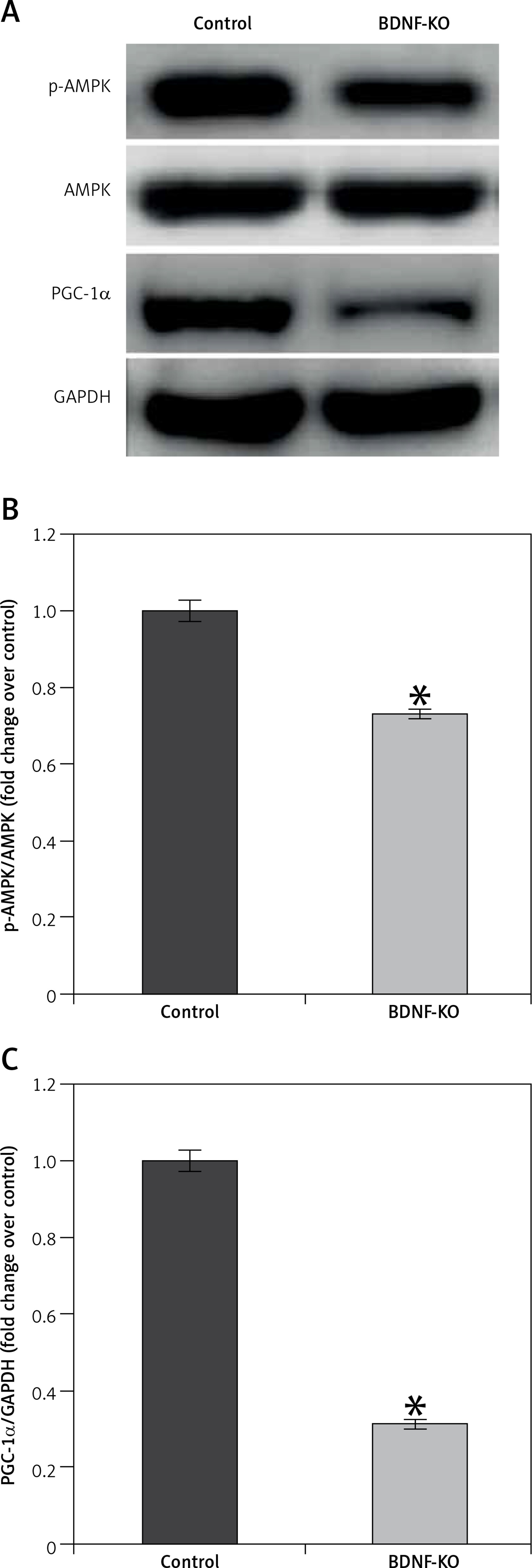
To clarify the role of BDNF in the maintenance of exercise capacity, we examined the effects of its depletion in C2C12 myoblasts. BDNF siRNA resulted in a markedly decreased level of phosphorylation of AMPK and PGC1α proteins (Figure 5 A). A control siRNA had no such effects. These results suggest that BDNF may regulate skeletal muscle function in HF mice after MI through AMPK-PGC1α signaling.
Figure 5
P-AMPK and PGC-1α expression in untreated and BDNF siRNA treated C2C12 myoblasts. A – Representative blots of p-AMPK and PGC-1α. B, C – Relative protein band density values of p-AMPK and PGC-1α were calculated as the ratio of protein of interest to that of GAPDH
Data are presented as means ± SE. *P < 0.05 vs. Control group.
Discussion
In the present study, we demonstrated for the first time that restoring the BDNF/TrkB and AMPK/PGC-1α signaling pathway is a crucial mechanism by which exercise training attenuates muscle dysfunction in HF. Our main finding was that exercise training significantly attenuated the reduction in the expression of BDNF, p-TrkB, p-AMPK and PGC-1α, restoring exercise tolerance.
A wealth of evidence has indicated that regular exercise can protect individuals from a host of cardiovascular and metabolic diseases [3, 26]. Indeed, exercise training is emerging as a key intervention for preventive cardiology [27] and a nonpharmacological strategy for HF therapy [28]. Such relevance is due to the ability of exercise training to counteract neurohumoral overactivity in HF [24, 29], which attenuates cardiac and skeletal muscle dysfunction [5, 30], and ultimately leads to improved exercise tolerance and life quality in HF patients [31]. However, the mechanisms by which exercise training can prevent cardiovascular diseases are not completely understood. In the present investigation we demonstrate that exercise training in MI-induced HF mice increased BDNF, p-TrkB, p-AMPK and PGC-1α levels, and prevented skeletal muscle atrophy and cardiac dysfunction.
BDNF plays a key role in neuron development, survival, and function, and seems to play a key role in mediating the benefits of exercise [32, 33]. BDNF is shown to be produced in the skeletal muscle, and increased by muscle contraction to enhance fat oxidation, which can regulate glucose and fat metabolism [34, 35]. The decrease in BDNF levels may be due to the physical inactivity in HF patients, and could impair the metabolism of skeletal muscle and lead to exercise intolerance [36, 37]. Furthermore, BDNF levels in the skeletal muscle can be enhanced by exercise training [38, 39], and a beneficial effect of exercise training on HF might be due to BDNF release from the skeletal muscle. Lee et al. found that BDNF is induced by exercise training in skeletal muscle and the non-infarct area of the LV, which may contribute to improvement of muscle dysfunction and cardiac function after MI [40]. Exogenous BDNF increases evoked acetylcholine (ACh) release at the neuromuscular junction (NMJ) and the TrkB receptor is normally coupled to this process [41, 42]. These results suggest that BDNF may play a critical role in metabolic derangements [35]. Further studies are needed to better understand the pathophysiology underlying the relationship between BDNF levels in skeletal muscle and exercise intolerance in HF. Our data reinforce the reduction in BDNF and p-TrkB protein levels in skeletal muscle of HF mice. Here we report that the mechanisms underlying the restoration in exercise tolerance and amelioration in ventricular function include the prevention of metabolism abnormalities by changing the BDNF level and phosphorylation status of TrkB proteins.
Peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) is a powerful stimulator of mitochondrial biogenesis and gene transcription in the skeletal muscle [19]. AMP-activated protein kinase (AMPK) is an upstream regulator for PGC-1α [43], mediating PGC-1α transcriptional activation in response to various stimuli [44]. Regarding downstream BDNF/p-TrkB signaling pathway, we observed that exercise training increased PGC-1α expression levels and re-established AMPK phosphorylation levels in HF mice. Increased p-AMPK and PGC-1α levels suggest a major role of AMPK/PGC-1α in the anabolic effect of exercise training when muscle dysfunction is taking place. This is an important result, highlighting the homeostatic role of exercise training in counteracting muscle dysfunction rather than increasing AMPK/PGC-1α signaling that is known to be activated by exercise training in eutrophic conditions [45].
These results suggest that the mechanism underlying exercise-induced cardioprotection and maintenance of exercise capacity may be influenced by factors such as training regimen and HF etiology. Our study shows that in a model of MI induced HF, exercise training restored BDNF, p-TrkB, p-AMPK and PGC-1α expression to control levels. One may consider that exercise training favors mitochondrial biogenesis by AMPK/PGC-1α, preventing the impairment of metabolism by BDNF. Herein, we demonstrated that improved muscle function is associated, at least in part, with a reestablished BDNF/TrkB and AMPK/PGC-1α signaling pathway.
In conclusion, our findings provide strong evidence for the contribution of BDNF to exercise training maintaining exercise capacity and regulating cardiac function in HF mice after MI through AMPK-PGC-1α signaling. Altogether, these findings strengthen exercise training as an efficient therapeutic approach in HF. Future studies should investigate the ideal exercise intensity and duration in order to optimize both muscle anti-atrophic effects of exercise training in HF.