The mainstay of clinical care for patients with NF-1 and related rasopathies
Neurofibromatosis type 1 (NF-1) is one of the most common single gene disorders in the global population [1, 2]. It is a primary neoplasia disease, as one of two fundamental diagnostic symptoms is neurofibromas, present in almost all patients [1, 3–6]. The main diagnostic sign presented in almost all patients from birth in a continuously growing and significant number is multiple café au lait spots (CALs): flat and superficial hyperpigmented hairless spots, significantly distinct from congenital and acquired melanocytic nevi, with no propensity for malignant transformation, characterized by a linear border [7]. A diagnosis of NF-1 is still based on clinical criteria developed in 1988 (NIH-CC-88) [8], before identification of the Nf1 gene and its product in 1991 [9], presented in Table I (bolded points 1–7). NIH-CC-88 does not include the pathognomonic sign of NF-1 such as the presence of T2 hyperintensities on brain magnetic resonance imaging (MRI), which are non-tumoral white matter lesions, referred to as unidentified bright objects (UBO) or FASI (focal areas of signal intensity) [1, 6, 10–12].
Table I
Diagnostic criteria and clinical characteristics of neurofibromatosis type 1 and related RASopathies
| Disease | Diagnostic criteria (patients’ frequency) | Other frequent symptoms | Other symptoms not frequently observed in patients |
|---|---|---|---|
| NF-1 | 1. Six or more café-au-lait macules over 5 mm in diameter in prepubertal individuals and over 15 mm in greatest diameter in postpubertal individuals CALs (> 98%) | Kyphoscoliosis at age of puberty (bone dysplasia) Ligament apparatus defects and hypermobility of joints Short stature or failure to thrive Macrocephaly Hemodynamically meaningless heart defects (mostly valvular) Large arteries aneurysm, mostly renal artery stenosis complicated by arterial hypertension, Willis cerebral arterial circle malformations and Moyamoya disease Speech delay and functional deficits Learning and cognitive problems (up to 75% in school-age but less significantly apparent in adults) | Intellectual impairment, mild (< 20%) ADHD-like syndrome (app. 38%) Precocious puberty/menopause (but delayed as well) Brain WHO/IV astrocytomas (glioblastomas) (seldom) Non-malignant tumors of spinal cord (unique) Primary arterial hypertension Malignancies (general long life risk approx. 5%): |
| 2. Axillary or inguinal freckling (70% in school ages and 85% in adults) | |||
| 3. – 2 or more skin NFM (> 98%); perispinal NFM* – 1 or more superficial and/or deep PNF (30% and 50%)* | |||
| 4. 2 or more Lisch nodules* (90–95%) | |||
| 5. Bone dysplasia (sphenoid wing or long-bone in infancy)* | |||
| 6. OPG (15–20% in early childhood; stable since adolescence) | |||
| 7. First-degree relative (parent, sibling, or offspring) with NF-1 diagnosed by the above criteria | |||
| Brain UBOs observed in NMR in about 60% of patients in childhood (not adult)** | |||
| sNF-1# | Skin manifestations indistinguishable from NF1 (CAL i NFM/NFS), but limited to one body region (e.g. only one extremity) | ||
| fs-NFⓍ | Isolated spinal nerve root neurofibromas, symmetric and multiple, with or without CALs, Lisch nodules and(or) axillary or inguinal freckling (no other S&S of NF-1) | ||
| NFLS (LS) | Multiple CALs characteristic for NF1 Macrocephaly (head circumference > 95 percentile) Less severe than NF-1 and not associated with neurofibromas, OPGs, Lisch nodules | Axillary or inguinal freckling (less frequently than in NF-1) UBOs in NMR (seldom) Often pulmonary valve defects (or other minor heart defects, mostly valvular) Multiple lipomas (benign) Variable dysmorphic features | Learning and cognitive problems and intellectual impairment (more frequent than in NF-1); speech delay and functional deficits, ADHD-like syndrome Vascular malformations Meaningless malignancy risk (not definitely proved) except ANLL (as for NF-1) |
| NFNSⓍ | Consolidated phenotypes of both diseases:
| Characteristic NFM with less frequent occurrence of PNF and lower risk of malignancy | |
| JCSⓍ | Complex of multiple nonossifying fibromas of the long bones, mandibular giant cell tumors, and cafe-au-lait macules in individuals without neurofibromas | Learning and cognitive problems and intellectual impairment as well as hypogonadism or cryptorchidism are frequently observed with less often ocular and cardiovascular malformations | |
| WSⓍ | Characterized by CALs and axillary or inguinal freckling together with pulmonic stenosis, decreased intellectual ability and short stature | Most affected individuals have relative macrocephaly and Lisch nodules and about one-third of those affected have neurofibroma | |
CALs – cafe-au-lait spots, NFM/PNF – neurofibroma and plexiform neurofibroma, OPG – optic nerve glioma, MPNST – malignant peripheral nerve sheath tumor, JMML – juvenile myelomonocytic leukemia, ANLL – acute non-lymphoblastic leukemia(s), RMS/STS – rhabdomyosarcoma/soft tissue sarcoma, GIST – gastrointestinal stromal tumors, BRCA – breast cancer, PhCC – pheochromocytoma, DCT – duodenal carcinoid tumor, sNF-1 – segmental NF-1, fsNF – familial spinal NF, NFLS(LS) – NF-like syndrome (Legius Sy.), NFNS – neurofibromatosis-Noonan syndrome, WS – Watson syndrome, JFC – Jaffe-Campanacci syndrome.
Inherited as an autosomal dominant trait, the NF/RAS group includes NF-1 and its allelic forms resulting from germline mutations or gross deletion of the Nf1 gene (locus 17q11.2) together with Legius syndrome (NF-like syndrome, NFLS), caused by mutations in the SPRED-1 gene (locus 15q13.2) (Tables I and II) [1, 4, 6]. Both genes are tumor suppressors of the Ras-MAP-kinase signal transduction pathway and have profound effects on cell development and growth control [5, 13]. Thus NF-1 (but less significantly its allelic forms and NFLS) exposes carriers to higher risk of malignancy than the general population (Table III) [3, 5, 14, 15].
Table II
Epidemiologic characteristics of neurofibromatosis type 1 and related RASopathies
Table III
Risk of neoplasia in NF-1
| Type of malignancy | Lifetime risk of given neoplasia (as a definite percentage or in comparison to general population risk) |
|---|---|
| Superficial neurofibromas (NFM) | > 98% |
| Plexiform neurofibromas (PNF) | 30–50% (superficial or deep) |
| Optic nerve glioma (OPG; WHO I) | 15–20% |
| Malignant peripheral nerve-sheath tumor (MPNST) | 6–10% |
| Leukemia (mostly non-lymphoblastic – ANLL, but especially juvenile myelomonocytic leukemia – JMML*) | Exceeds population risk 7-fold* |
| Brain tumors (CNST) (mostly WHO I/II astrocytoma/glioblastoma) | Exceeds population risk 5-fold |
| Rhabdomyosarcomas (RMS) (and other soft tissue sarcomas – STS) | 1.4–6% |
| Gastrointestinal stromal tumors (GIST)** | 4–25% |
| Breast cancer (BRCA)** | Exceeds population risk 5-fold |
| Pheochromocytoma (PhCC)** | 0.1–5.7% |
| Duodenal carcinoid tumor (DCT)** | 1% |
| General estimated long-life risk of cancer | Approx. 5% |
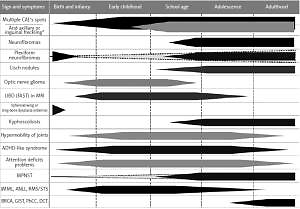
Neurofibromatosis type 1 with 100% penetration of the gene mutation by the age of 20 observed in 1 out of 2500–3000 live births, outstanding clinical heterogeneity between patients even in one family and a weak genotype-phenotype correlation, a high rate of spontaneous mutation exceeding 50%, remarkable age-dependent onset of different disease signs and symptoms (Figure 1), and multiple comorbidities with the most important increased risk of malignancy, warrants unique multispecialty complex medical care, separate from remaining RASopathies and other phacomatoses [1, 3–6, 15]. Furthermore, the physician faced with a toddler presenting multiple CALs only, whose parents had no NF-1 diagnosis, have to differentiate NF-1 among approximately 80 clinical entities described in Online Mendelian Inheritance in Man [1, 4, 6, 16, 17].
Figure 1
The age of presentation and occurrence of the most frequent or the most important signs and symptoms of neurofibromatosis type 1 CALs – cafe-au-lait spots, MPNST – malignant peripheral nerve sheath tumor, JMML – juvenile myelomonocytic leukemia, ANLL – acute non-lymphoblastic leukemia(s), RMS/STS – rhabdomyosarcoma/soft tissue sarcoma, GIST – gastrointestinal stromal tumors, BRCA – breast cancer, PhCC – pheochromocytoma, DCT – duodenal carcinoid tumor. *Currently assumed as a continuum, not separate sign.
Currently, newer molecular tools based on microarray analysis of gene expression at different levels of the transcription process (genomics, proteomics and metabolomics) supported by large-scale data analysis statistics provide the possibility to find yet undisclosed genotype-phenotype correlations, improve epidemiology or allow reliable and quick differential diagnosis among those 80 clinical entities (or more if new diseases are discovered due to those analytical tools) [18, 19].
Children from birth until 5th year of life
Multiple CALs are almost an obligatory sign of NF-1 [1, 3, 4, 6, 10]. In toddlers whose parents had no NF-1 (approx. 50% of patients) there are precisely 3 clinical entities allowing certain diagnosis of NF-1: (1) child with multiple CALS and (or) (2) tibia or sphenoid wing dysplasia and (or) (3) inborn visible plexiform neurofibroma (PNF). Since delivery, PNF may be stable or fatal with tremendous disfiguration, especially when growing within the head and neck area (3–5% of patients until 5th year) [1, 6, 20]. Congenital tibial dysplasia (CTD) often resulted in pseudarthrosis and is a devastating sign of NF-1, observed in 2% of children up to 3rd year [1, 6, 21, 22]. Many children affected with CTD become disabled and required repeated reconstructive surgery, especially by longstanding Ilizarow’s modality [22]. Until now promises from preclinical experiments with genetically modified Nf1 mice have failed in clinical trials performed in humans [23, 24]. A separate problem is devastating and inborn sphenoid wing dysplasia observed in < 1% of neonates, especially when accompanied by facial PNF [25]. As the above-described signs are seldom present at this age, the vast majority of Nf1 spontaneous mutation toddlers are too young to fulfill NIH-CC-88 (Figure 1). Nevertheless, NF-1 may be suspected with a high probability when they present more than 10 indisputable CALs at the 3rd, or more than 6 below the 2nd year only [26, 27].
The main criticism toward NIH-CC-88 concerns a risk of undisclosed optic nerve glioma (OPG) in young children without yet confirmed NF-1 diagnosis [28, 29]. The OPGs are disclosed in MRI in approximately 15–20% of patients up to the 7th year, but from this age are characterized by a stable or regressing course. Its prevalence in older children and adults is unique [29]. Only approximately 5% of them are symptomatic and may require treatment preserving future vision impairment, seen mostly in ophthalmological investigation as a narrowing of the visual field [1, 14, 29]. In case of orbital disfiguration, and/or rapid progression, and/or clinically significant impaired vision chemotherapy is a mainstay of treatment (coordinated by Polish Pediatric Neurooncology Working Group). In children with NF-1 and OPG surgery is contraindicated [1, 14, 30]. In this age MRI obligatorily requires general anesthesia and should not be done routinely and permanently in every child with multiple CALs and no signs of NF/RAS (see later), besides a few exception [11, 14]. Apart from OPGs the other important indication for cerebral MRI is macrocephaly, especially progressive, observed in 1.5 % of toddlers with NF-1 aqueduct stenosis and/or hydrocephaly. Sporadic, not NF-1-dependent brain tumor may arise as well [1]. In toddlers presenting developmental delay MRI should always be considered, but routinely not earlier than in the 3rd year. In contrast, children with multiple CALs (but all presenting secondary regression in developmental milestones) may require early “prophylactic” MRI when tuberous sclerosis (TSC) is suspected. It is important, as the prophylaxis of regression in psychomotor development or severity and the time of epilepsy occurrence is possible in these patients [31]. On the other hand, the unjustified fear of TSC and possible but unlikely NF-1 dependent brain tumor as well as weak experience in pediatric oncology among neurologists lead to unintentional malpractice when NF-1 children are admitted to neurologists instead of NF/RAS centers. Epilepsy (approximately 8% of children) and severe neurological problems are uncommon in NF-1 [1, 4] whereas tumor derived neuropathic pain and other neuropathic sensations (swelling, hyper- or hypoalgesia, paresthesia or paresis) require surgical (neurosurgical) or oncological consultation and care. The role of the ophthalmologist is important in older patients with NF-1 but not toddlers as both OPGs and hydrocephaly should be diagnosed clinically and confirmed on imaging long before any ophthalmological disclosures; yet visual field examination in children is reliable from the 8th to 12th year [1, 4].
The physical and sensorimotor development of neonates with NF-1 is usually proper, but at the end of this period some children do not follow sequential skill in learning to talk and require logopedist support and supervision. From the 2nd year attention deficit and hyperactivity may be observed [1, 4].
Skin juvenile xanthogranulomas is more prevalent in toddlers with NF-1 than in the general population [32]. Appearing between the 2nd and 6th year, usually self-limiting and disappearing within 1 to 3 years from symptoms occurring, and meaningless in a majority of healthy children, in NF-1 toddlers it has been linked to a possible increased risk of leukemia, although this association is not totally compelling [33]. Occasionally systemic xanthogranulomatosis presents a risk for any child, as it resembles Langerhans cell histiocytosis with all its clinical and life-threatening consequences [34].
Children from the 6th to the 12th year of age
From the 6th year, NF-1 becomes a more distinct disorder, as two other characteristic and common signs appears: axillary and inguinal freckling, usually detected in approximately 70% of affected individuals by the age of 5–8 years (85% of adults), and Lisch nodules, which are benign melanocytic hamartomas of the iris, typically first noticed in children aged 5–10 year and found in 90–95% of adults with NF-1 [1, 4, 6]. Lisch nodules usually precede the appearance of cutaneous neurofibromas (NFM), which are the third relevant NF-1 diagnostic signs [1, 6]. There are four subtypes of NFM: cutaneous, subcutaneous, nodular or diffuse plexiform, and spinal [3, 4].
Intracutaneous NFM often occurs as pinkish-purple, raised, soft lesions that can then transform into more “wart”-like growths, but never undergo malignant transformation. Local pruritus may occur and substantial discomfort or disfigurement may arise, when they are present in hundreds or thousands. Routine surgical excision is still controversial. Tumor removal should be reviewed when pain and functional deficits occur or growth as significant exophytic masses with a gravity effect of “sagging” is observed, but the decision should always be weighed up according to a risk-benefit assessment (high risk of neurological complication versus regrowth) [35].
Spinal NFMs can occur at single or multiple nerve roots (Christmas tree sign) and might be associated with both sensory and motor deficits. They may also cause tumor related nondystrophic kyphoscoliosis. Despite being localized and isolated, familial spinal NFM (fsNF) is assumed as an allelic form of NF-1 [36].
Occult PNFs represent an early embryonic origin tumor and may be present even in the fetus, but become apparent in infancy or enlarge later on, most prominently during the second decade of life. They develop finally in about 30% (visible) to 50% (on imaging) of individuals up to the 18th year [1, 4]. In contrast to superficial NFM, deep PNF arise from multiple nerve fascicles and grow along the length of a nerve infiltrating surrounding structures, causing substantial pain and even neighborhood bone destruction. Although the best therapeutic option for symptomatic lesions is surgical removal, this approach is quite often technically impossible because of severe complications, mostly functional [35].
Deep internal PNFs present a lifetime risk of malignant transformation into malignant peripheral nerve sheath tumor (MPNST), reaching approximately 8% [1, 3, 5]. Malignant transformation is uncommon in childhood, but occurs in adolescence and adulthood. When MPNST diagnosis is confirmed in a biopsy, the patient requires aggressive treatment according to the Polish oncological standard of complex soft tissue sarcoma therapy (currently of CWS-Study-Group), starting with neoadjuvant chemotherapy. The only controversial element of treatment is radiotherapy, when other, not yet transforming PNFs are located in the irradiation field (risk of provoked malignant transformation) [1, 4, 5]. Besides the tremendous attempt made to find an effective treatment of PNFs, including biologically targeted therapies, to date no cure exists, except rare cases of possible radical surgical excision and selumetinib [37], the first product assigned for treatment of PNF in NF-1, registered in US by the FDA as orphan drug in 2018 [38], which received orphan designation from the EMA in 2018 as well [39]. Disappointment concerning other miracle medicines used previously still warrants skepticism toward any unproved therapy.
The most important complications of NF-1 in school age children are deficits in cognition and behavior, presenting mostly as attention deficit and hyperactivity together with delays and problems of speech, as well as learning difficulties [40]. Neuropsychological deficits include a lowered average IQ (intellectual impairment of IQ < 70 affects < 20% of patients), as well as academic (75–80%) and visuospatial skills problems, social competence, and attention paucity [40, 41]. These require special education and/or remedial teaching [42]. Groups at risk are boys and children with lower verbal IQ and ADHD-like behavior (approximately 38%) [40]. Promises from preclinical experiments with genetically modified Nf1 gene mouse have not been confirmed clinically so far [43, 44].
Symptomatic hydrocephaly at this age usually indicates WHO I/II cerebral astrocytoma (glioblastoma), observed in 2–3% of patients with a lifelong risk of occurrence [14]. Other brain tumors present almost populational prevalence [4, 14].
Endocrinopathy and growth disturbances are multifactorial or idiopathic, and sometimes require growth hormone therapy [1, 4].
Adolescence and adulthood
The complications of NF-1 appearing in adolescents and adults comprise: (1) growing risk of malignancy (Table II) [5, 45], (2) endocrinopathy (mostly precocious or delayed puberty and menopause; thyroid dysfunctions), (3) posture bone dysplasia together with joint hypermobility resulting in dystrophic (kypho)scoliosis, additionally complicated by paraspinal NFMs deteriorating spinal column statics and morphology [21, 35, 36], (4) development of cardio, cerebro- and renovascular abnormalities with hypertension and aneurysms, and (5) many other, rare NF-1 comorbidities [1, 4, 6, 10]. Apart from the known risk concerning transformation of PNF into MPNST, gastrointestinal stromal tumors, soft tissue sarcoma or cancers [5, 45], recently a conjunction between NF-1 and moderate risk of poor prognosis breast cancer (BRCA) has been disclosed [46].
Standard imaging examinations [1, 4–6, 11, 12, 14, 15, 21]
The examination preferred to assess NF-1 associated morbidities depends on the clinical course and the organ involved. A gold standard is both standard and contrast-based ultrasound for either screening or diagnostic approaches. Except other causes, cranial sonography in toddlers with NF-1 is dispensable, especially when posterior fossa arterial malformations and tumors are elusive for it. As a screening method, abdominal USG should be performed in childhood at least once a year during a routine NF visit. The indication and frequency of remaining fields USG, mostly tumor-directed, should depend on the course and severity of morbidity. Echocardiography should be done at least once in each toddler, as a mainstay of differential diagnosis of a “child with multiple CALs” (e.g. NFLS, TSC or other RASopathies).
A plain radiograph is currently reserved for skeletal lesions (e.g. long bone dysplasia or tumors, especially non-ossifying fibroma) or measures of Cobb’s angle in case of NF-1 related (kypho)scoliosis and as an assessment of bone age (in case of precocious or delayed puberty of McCune Albright syndrome) and structure of hand (e.g. thumb deformation in differentiation with Fanconi’s anemia). Yet, in some instances it is currently being replaced by USG to prevent recurrent irradiation.
Non-routine MRI is the investigation of choice for precise diagnosis and longitudinal observation of complicated cases (mostly tumors and spine), FASI and OPGs or vasculopathy. In Polish practice the brain and orbital MRI is performed even in an asymptomatic child at least once in the 3rd year and then repeated routinely until the 15th–18th year, not more often than every 1.5–3 years, unless otherwise necessary (progression of tumor in previous imaging or clinical symptomatology). In adolescents and adults brain MRI is required only when the patient presents symptoms. The MRI of other regions, especially spinal, is required in response to clinical presentation of NF-1 complication, but not routinely. Whole-body MRI is a reference standard to identify and follow the dynamics of the tumors in chosen patients. When malignant transformation of PNF into MPNST is considered, the precise contrast MRI, and dynamic MRI or PWI, DWI, NMR spectroscopy as well, may be of utmost importance. Whereas FDG PET/CT has almost 100% sensitivity in this indication, the specificity is assumed as 40% in asymptomatic [47] to 77–95% in symptomatic cases [48], but with a well-known limitation and low positive predictive value < 25% [47–49] and a probability of false positive results in PNFs. Any imaging proved suspicion of malignant transformation requires multiple biopsies taken from the most biologically active parts of the tumor, even in the MRI guided modality.
Radionuclide scans are needed in pheochromocytoma (functional imaging) or as a staging procedure of MPNST, and rarely when multiple benign bone tumors are suspected (e.g. differential diagnosis of McCune-Albright syndrome).
According to the worldwide consensus CT scans should be avoided in NF-1 patients, because of irradiation risk and the irrelevant results (e.g. inability to disclose FASI) [1, 4, 6].
Molecular diagnosis (DNA analysis)
Even currently not every child requires routinely molecular testing, as a majority of them are diagnosed according to NIH-CC-88 criteria together with MRI before the 4th year [6, 10]. Molecular analysis does not improve care and has almost no predictive impact as the weak genotype-phenotype correlation and extreme clinical heterogeneity of NF-1 is observed [1, 4, 16, 50–52]. Testing is necessary only: (1) when NFLS or constitutional mismatch repair deficiency syndrome (anamnesis!) is suspected; (2) in a suspected toddler with a serious tumor in whom diagnosis of NF-1 immediately affects management (e.g. in OPGs), or (3) if prenatal or preimplantation genetic diagnosis in a current or future pregnancy is anticipated in an adult with clinical diagnosis only [52]. The exception comprises at-risk relatives of fsNF-1 or identified NF-1 carriers of the c.29702972-delAAT pathogenic variant, whereas affected individuals from the rest of the family may not ever meet the NIH criteria [53].
Four types of large Nf1 deletion [54] and the other three specific genotypes as well as increased missense mutation rate in fsNF [55] have been identified to date. A mild NF-1 phenotype characterized by CALs and skinfold freckling but not visible neurofibromas results from the recurrent three base-pair in-frame deletion within Nf1 exon 17th (c.2970-2972 delAAT) [53]. Well-established genotype–phenotype correlation is associated as well with missense mutations affecting codon p.Arg1809, where patients with Noonan-like features observed in > 50% (pulmonic stenosis, short stature, developmental delay) exhibit CALs and Lisch nodules, but no visible NFM/PNF [56]. Severe phenotype results from deletions and just recently described missense mutations affecting Nf1 codons 844–848 [57].
The newer microarray molecular tools which allow more precise and fast confirmation of disease due to the complex large gene expression and methylation data profiles utilizing the large-scale data analysis statistics, are currently promising in diagnosis of the most complicated cases [18, 19].
A model of coordinated care for neurofibromatosis type 1 and related rasopathies in Poland
Disease management defined as “a system of coordinated healthcare interventions and communications for populations with conditions in which patient self-care efforts are significant” applies not only to patients with common chronic illnesses, but may be adapted to the field of rare diseases [58, 59]. The pediatric definition of care coordination given by the American Academy of Pediatrics in 2005 stated that “it is a process that facilitates the linkage of children and their families with appropriate services and resources in a coordinated effort to achieve good health” [60]. Thus, the system of coordinated medical care (CMC) offered to patients suffering from NF/RAS in Poland combines complex multispecialty consultation with permanent supervision and patient’s oriented longitudinal care and provides a sense of medical security to the patients with chronic and devastating illness. It is provided at Pediatric Oncology Departments of Medical Universities in Warsaw and Bydgoszcz based on a trusted CMC model entity verified by more than 14 years’ experience. The general idea is to ease the incurable and chronic disease path which has an additional impact on direct and indirect health care expenses, which might be significantly reduced, and systemic medical resources, which can be provided more efficiently [59].
The described complex health problems of NF-1 definitely require multispecialty consultation or morbidity-oriented care provided by an NF/RAS reference center [3, 10, 50]. Nevertheless, the compound complexity of care warrants the whole process of care longitudinal coordination as well. It refers to a regular yearly clinical visit at the NF/RAS Center based on ambulatory consultation and ability to consult forthcoming patient’s morbidity both at a visit “on demand” or throughout electronic media (Table IV) [51]. It is provided by a “medical reviewer” and an expert in disease management (called the “NF-coordinator”) supervising patient-oriented care (Figure 2). An external facility provided by a center of excellence experienced in highly sophisticated surgery or care is seldom required. An example of such may be peripheral nerve [61] or face transplantation (done for the first time in Poland in 2016) [62]. Some patients may need both specialized treatment in an external center of excellence (e.g. treatment of OPGs or NF-1 related CNS tumors in Polish Reference Center for Brain Tumors) together with permanent care of the NF/RAS-CMC.
Table IV
NF/RAS Coordinated Medical Care responsibilities in Poland
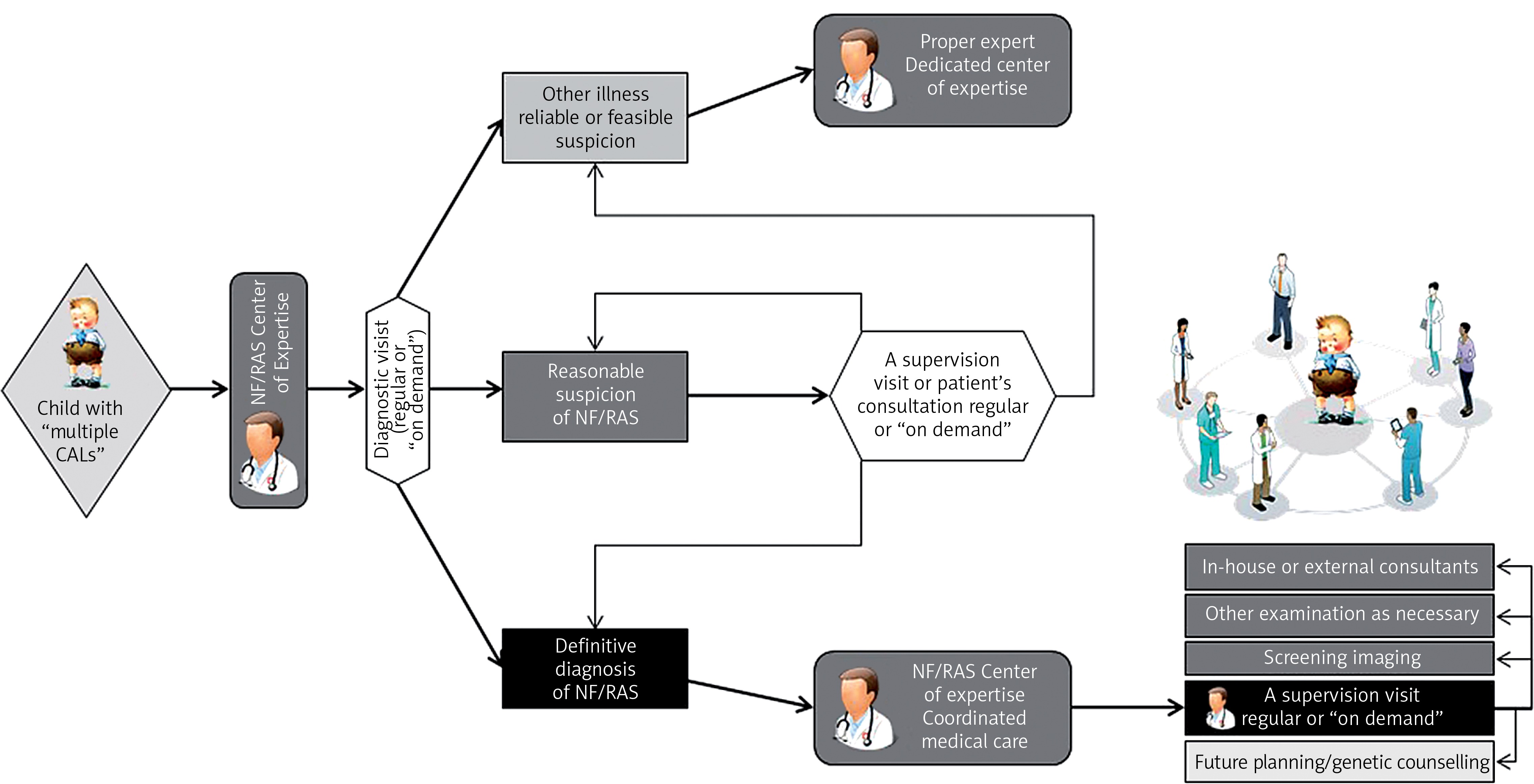
The NF/RAS reference center responsibility is not only the permanently supervised monitoring and holistic management of disease together with education and psychological support of the patient and family, but differential diagnosis of NF-1 as well, troublesome because of its complexity and age-dependent manner as well as sparse symptoms of the diseases in toddlers. The center is additionally obliged to provide a forum for clinical audit and academic interaction throughout the multidisciplinary meetings and telemedical conferencing and for permanent interactions with parental organization.
Conclusions
The mainstay of NF-1 management is patients’ oriented, both multispecialty and coordinated care, comprising age-specific monitoring of disease manifestations and comorbidities treatment as well as patient education, which requires a diversified attitude towards children at different ages, continuously supervised by the NF coordinator. The coordinator is responsible for the patient’s directed, holistic management providing a sense of medical security to the patient and family. Such a system of care ended the socalled “diagnostic odyssey” of the patient’s family, hopelessly searching for proper and professional medical advice.