Introduction
Focal and segmental glomerulosclerosis (FSGS) is an important cause of steroid-resistant nephrotic syndrome in human beings, the pathogenesis of which, however, has not been fully elucidated. Previous studies of familial FSGS cases in the past two decades suggest that inherent defects in the structure and function of podocyte are closely associated with FSGS [1]. As a podocytopathy, podocyte depletion and changes of the functional integrity play key roles in the initiation and progression of FSGS [2].
Located in the Bowman’s capsule of the kidneys, podocytes wrap around capillaries of the glomerulus and determine the glomerular permselectivity [3]. The function of podocytes is highly dependent on their morphology, which features interdigitating foot processes among adjacent podocytes [3]. The actin cytoskeleton, an integral component of the foot processes in podocytes, is regulated by a number of proteins expressed in podocytes, including nephrin, NEPH1, podocin, CD2AP and α-actinin-4 [1]. Studies on familial FSGS showed that all these proteins played a role in podocyte functions [4, 5]. These findings suggest that the function of podocytes depends on the integrality of their cytoskeletal structure and slit diaphragm.
By using exome sequencing and further filtration in a recent study, we identified FAM40A (encoding a family with sequence similarity 40, member A) as the contributing gene for FSGS.
Previous studies reported that FAM40A is involved in the cytoskeletal organization of tumor cells and may be a new regulator of cytoskeletal organization, cell morphology and migration [6]. In addition, FAM40A was shown to be involved in cytoskeletal rearrangement by interacting with RP6-213H19.1, STRN, PDCD10, TRAF3IP3, STRN3, PPP2R1A, MOBKL3, CTTNBP2NL, STK24 and PPP2CA [7].
Therefore, we hypothesized that FAM40A may play a role in the podocyte cytoskeleton and is associated with familial FSGS. In familial FSGS, mutant FAM40A may induce podocyte dysfunction by affecting cytoskeletal rearrangement. To determine whether FAM40A affected the cytoskeletal organization and cell morphology in mouse podocytes (MPs), we firstly demonstrated the localization of FAM40A, and then focused on F-actin change in depleted FAM40A and mutant FAM40A cells.
Material and methods
Cell culture
Conditionally immortalized mouse podocytes (MPs), a gift from Dr. Zhihong Liu, Nanjing University (Nanjing, China), were culture, maintained or differentiated as described previously. In brief, MPs were maintained in RPMI 1640 medium (Gibco, Life Technologies, USA) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 0.1 mg/ml streptomycin (Life Technologies). To propagate MPs, they were cultured at 33°C in permissive conditions in culture medium supplemented with 10 U/ml mouse recombinant γ-interferon (Life Technologies) to enhance the expression of temperature-sensitive large T-antigen. To induce differentiation, MPs were maintained at 37°C without γ-interferon in non-permissive conditions for 2 weeks.
Establishment of adenovirus vectors of RNAi, wild-type (WT) and mutant (MU) mouse FAM40A gene expressions
Three pairs of miRNA oligos were designed for the target gene FAM40A and connected with the pcDNA6.2-GW/EmGFP expression vectors to construct stable RNAi plasmids, from which the most optimal was pAd-EGFP-FAM40A-miR with an interference efficiency of 69% for knocking down the target gene FAM40A. Likewise, the second-generation adenovirus expression system pAd/CMV/V5-DEST was used to establish adenovirus that over-expressed FAM40A-WT/MU. Finally, silenced FAM40A, FAM40A-WT and FAM40A-MU over-expression models were established successfully (Supplementary Figure 1).

Immunofluorescence microscopy and image analysis
All procedures were performed at room temperature unless otherwise specified. MPs were grown on collagen-coated glass coverslips, fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min, washed with PBS for 3 min, permeabilized with 0.5% Triton X-100 in PBS for 5 min, and rinsed with PBS. The nonspecific binding sites were saturated in blocking solution (3% bovine serum albumin, and 0.02% NaN3 in PBS) for 40 min. Cells were then incubated with primary antibodies (pre-diluted in blocking solution) and stained overnight at 4°C. Secondary antibodies were diluted in blocking solution and incubated for 50 min. The primary and secondary control antibodies were stained simultaneously. F-actin was stained with 100 nM rhodamine-labeled phalloidin (Cat. # PHDR1 Lot # 053) for 30 min, and α-tubulin was stained with 1 μM paclitaxel, Oregon Green 488 conjugate for 120 min. Cells were stained with DAPI (1 μg/ml, Sigma-Aldrich, Gillingham, UK) for nuclear staining. Coverslips were mounted on glass slides using the commercial mounting medium GelTol. Specimens were visualized and photographed using a fluorescence microscope (OLYMPUS BX51, Japan).
To quantify actin assembly, transfected cells were randomly selected and F-actin fluorescence images were acquired with a fluorescence microscope. Cell membranes and membrane protrusions were outlined on the fluorescence image with a width of 5 μm (“membrane region”). The mean fluorescence intensity (mean pixel density) in the membrane region and remaining cytosol region was quantitated using Adobe Photoshop CS5. Fluorescence intensity was collected from 10–12 different cells per group.
RT-qPCR
mRNA was isolated from differentiated immortalized MPs using an RNA assay kit (Qiagen, Hilden, Germany) and transcribed in cDNA by reverse transcriptase. No reverse transcriptase was added in the negative control.
Immunoblotting
Cells were lysed in ice-cold buffer containing 1% Triton X-100, 125 mM NaCl, 10 mM Tris (pH 7.4), 1 mM EDTA, 1 mM EGTA, 2 mM Na3VO4, 10 mM sodium pyrophosphate, 25 mM NaF, and a protease inhibitor cocktail (Roche Diagnostics). After removing insoluble components by centrifugation (10,000 rpm) at 4°C for 10 min, the protein concentration of the supernatant was quantified using a commercial reagent (Bio-Rad). Equal amounts of protein were separated by SDS-PAGE under reducing conditions. Proteins were electrophoretically transferred to nitrocellulose membranes, blocked with 5% dry milk and incubated with primary antibodies (1 : 100 dilution) at 4°C for 12 h. After three washes, membranes were incubated with secondary antibodies conjugated with horseradish peroxidase, and signals were detected by Pierce ECL Western Blotting Substrate.
Results
Expression and localization of FAM40A in MPs
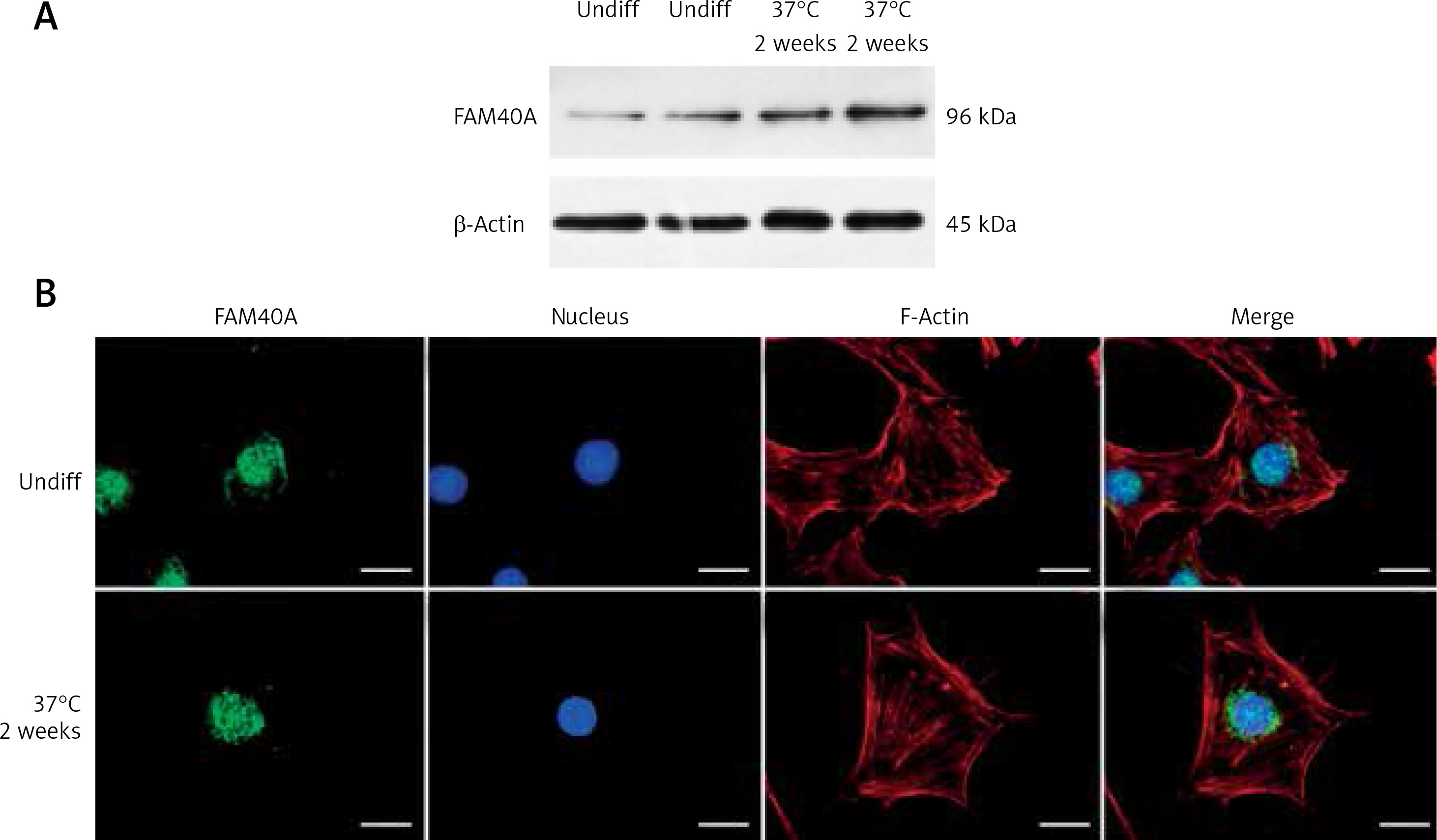
Characterized by marked proteinuria and podocyte injury, FSGS remains a challenge, and the alterations in structural genes of the podocyte contribute to the monogenetic forms of FSGS in the early stage [8, 9]. Podocyte-specific mutated genes, including NPHS1, NPHS2, WT-1, LAMB2, CD2AP, TRPC6, ACTN4 and INF2, have been shown to be responsible for FSGS [1, 10, 11]. However, the treatment options for those FSGS patients are limited, and steroid resistance has been an increased risk of dysfunction of the kidney [12–14]. Recently, we identified FAM40A as a contributing gene for familial FSGS by a genome-wide association study (GWAS, data not shown). To determine the role of FAM40A in podocytes, we first examined the expression of FAM40A in cultured MPs by RT-qPCR, immunofluorescence and protein immunoblotting. In immunoblotting, both undifferentiated and differentiated MPs showed the expression of FAM40A (96 kDa) (Figure 1 A). Expression of FAM40A in MPs was also confirmed by RT-qPCR. We next analyzed the localization of FAM40A in MPs by immunofluorescence staining. Based on the bioinformatic analysis of FAM40A, it was predicted to have three transmembrane domains binding to DNA [15, 16]. In undifferentiated MPs, FAM40A proteins showed nuclear localization with some enrichment on the nuclear membrane (Figure 1 B). Similarly, FAM40A had punctate localization mainly in the nucleus in differentiated podocytes (Figure 1 B). Remarkably, FAM40A was expressed and localized in podocytes, and significantly enriched in the nucleus and perinuclear zone (Figure 1 B).
Figure 1
Expression and localization of FAM40A in podocytes. A – Podocytes express FAM40A as shown by Western blotting from undifferentiated and differentiated MPs. B – Immunofluorescence staining of FAM40A in undifferentiated and differentiated MPs. FAM40A staining was concentrated in the nucleus with little cytoplasm around the protrusion. Scale bars = 10 μm
Altered expression of FAM40A in MPs disrupts the regular cytoskeletal organization and reduces the e expression of nephrin
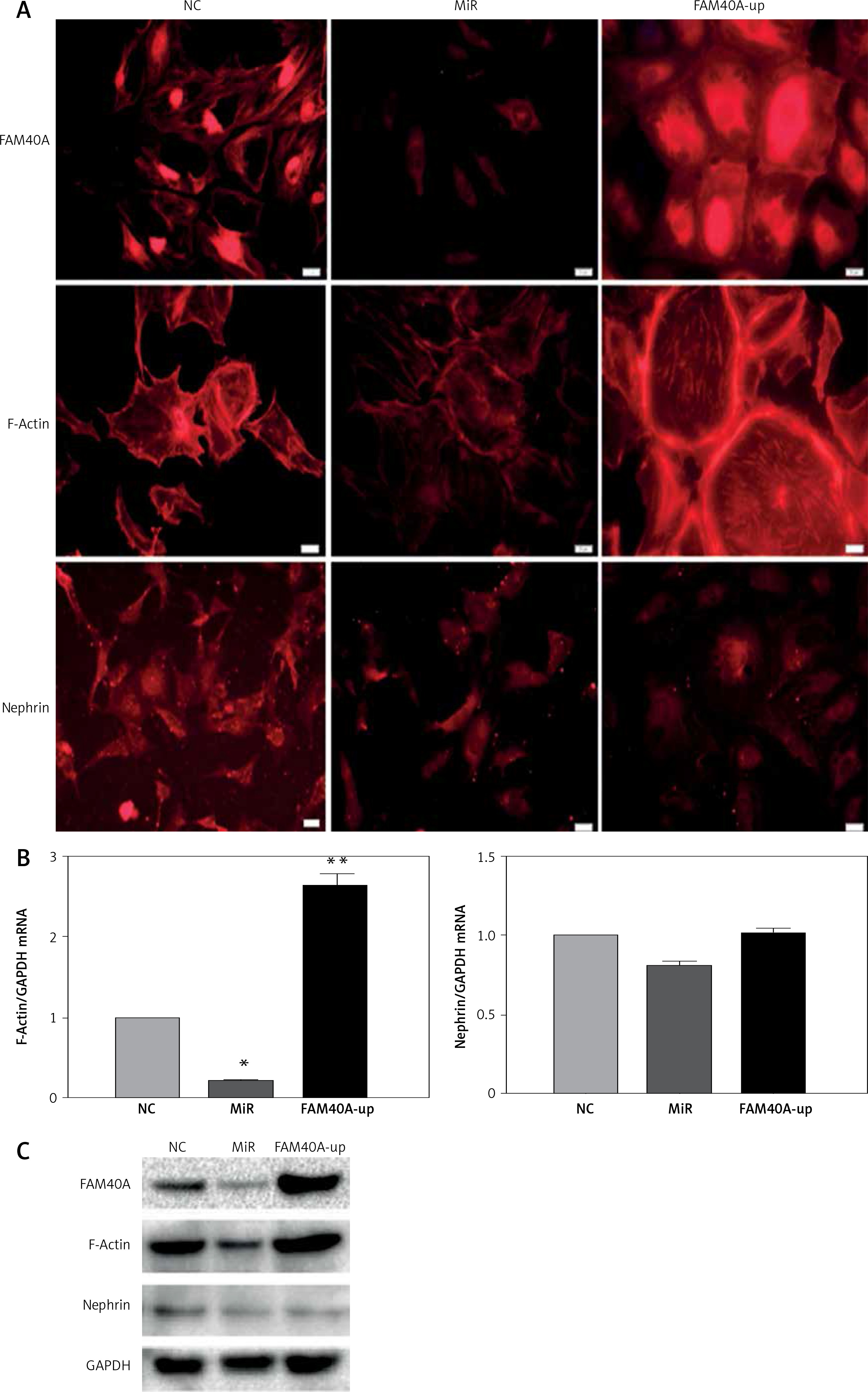
FAM40A has been identified previously in a novel large multiprotein assembly, together with FAM40B [7]. Moreover, FAM40A negatively regulates the MST3 and MST4 kinases and determines cancer cell migration and metastasis [17]. To explore the role of FAM40A in MPs, FAM40A-siRNA and FAM40A-overexpressed plasmids were transfected to fully differentiated mature MPs. Compared with the control group, MP morphology changed, and the normal foot-process organization disappeared when knocking down FAM40A. In addition, the intracellular expression of evenly distributed cytoskeletal protein F-actin was decreased (Figure 2 A), and the expression of F-actin was also decreased at both mRNA and protein levels (Figures 2 B, C). Moreover, the functional protein nephrin that was supported and fixed on the slit diaphragm by F-actin decreased (Figure 2 C). In contrast, MPs became larger and round, without the normal foot process organization, and the typical pseudopodial extension of foot processes disappeared (Figure 2 A). When the expression of F-actin protein was increased, its distribution was disrupted with obvious marginalization (Figure 2 A). Taken together, change in FAM40A expression altered the normal morphology and cytoskeletal architecture of MPs, mainly represented by disruption of F-actin distribution and disappearance of foot-process organization.
Figure 2
Altered expression of FAM40A in podocytes disrupts the regular cytoskeletal organization and decreases the expression of nephrin. A – Immunofluorescence staining of FAM40A depletion and over-expression on cytoskeletal organization in podocytes. Podocytes were transfected with the pAd-EGFP- FAM40A-miR or pAd-FAM40A-WT-EGFP in thin-bottomed multi-well plates, fixed for 72 h and stained for FAM40A, F-actin and nephrin. B – Detection of F-actin and nephrin mRNA by RT-PCR. C – Immunoblotting analysis of differentiated MPs
NC – negative control, MiR – interference group, FAM40A-up – FAM40A over-expression group. *Compared with NC group, p < 0.05; **compared with NC group, p < 0.01. Scale bars: 20 μm.
Mutation of FAM40A in podocytes disrupts F-actin and reduces the expression of nephrin
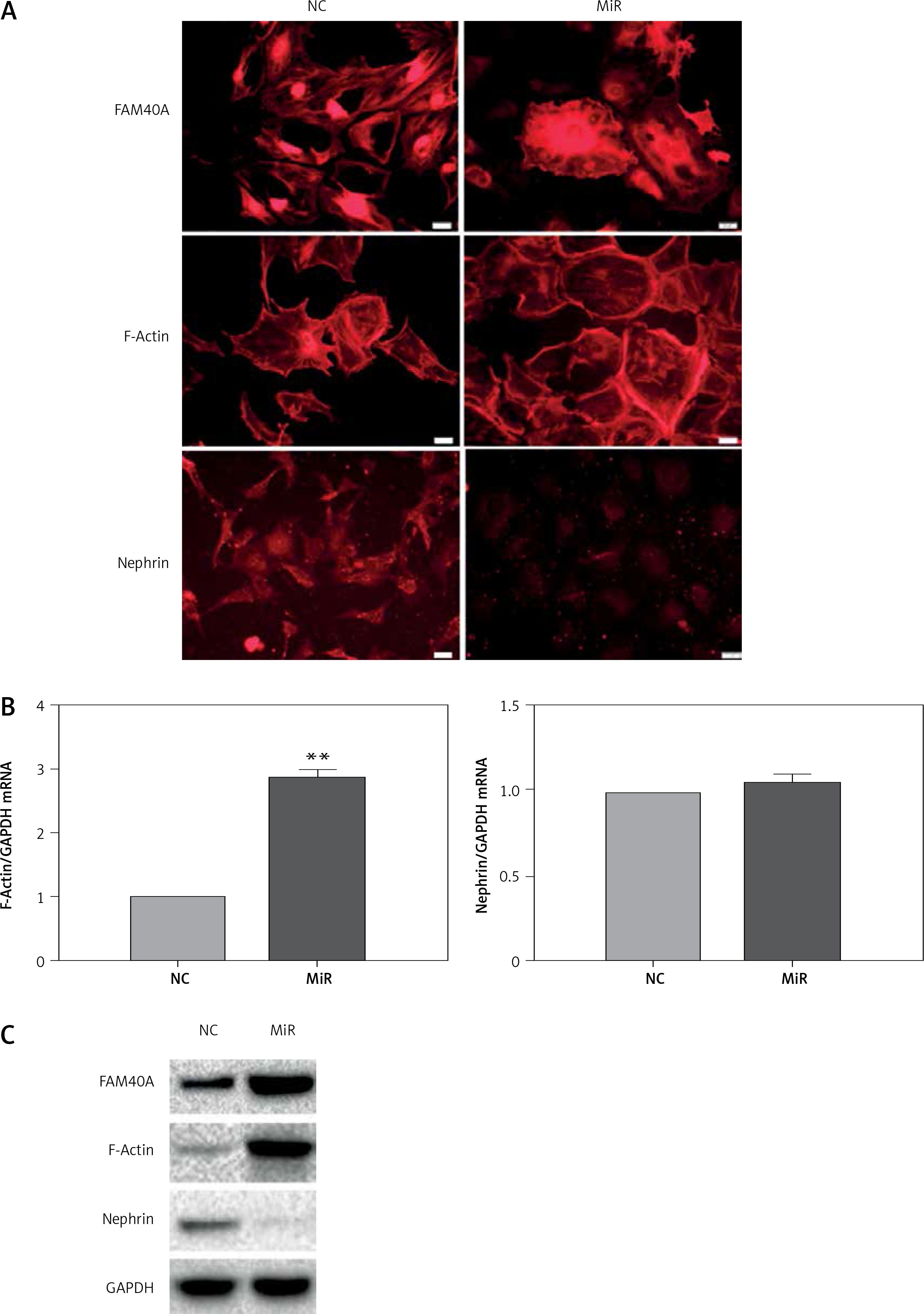
The mechanism by which FAM40A is involved in the progression of FSGS is largely unknown. To further explore the effect of FAM40A-MU on MPs, a plasmid harboring FAM40A-MU was transfected to fully differentiate mature MPs via the adenovirus vector. Fluorescence imaging showed that FAM40A-MU protein was expressed in MPs, and it changed the cell morphology of MPs, as represented by the wave-like irregular change of the cell periphery, and loss or fusion of the typical pseudopodia, extension of foot processes, increased marginalization of cytoskeletal protein F-actin distribution (Figure 3 A), and up-regulation of F-actin protein expression (Figure 3 C). When FAM40A mutation occurred, both immunocytochemistry and Western blot showed that the nephrin protein level was decreased, while nephrin mRNA expression remained unchanged (Figures 3 B, C). Taken together, FAM40A mutation induced morphological change of MPs. In addition, the expression of the functional protein nephrin of the slit diaphragm was decreased. Podocytopathy resulting from FAM40A-MU over-expression was similar to that from FAM40A-MT over-expression but it was not so dramatic.
Figure 3
FAM40A mutation in MPs disrupts F-actin and reduces the expression of nephrin. A – Immunofluorescence study of mutant FAM40A on cytoskeletal organization in MPs. MPs were transfected with pAd-FAM40A-MU-EGFP in thin-bottomed multi-well plates for 72 h, fixed and stained for FAM40A, F-actin and nephrin. Scale bars: 20 μm. B – RT-PCR mRNA of F-actin and nephrin. C – immunoblotting analysis of differentiated MPs
NC – negative control, MiR – interference group, FAM40A-up – FAM40A over-expression group compared with NC group, **p < 0.01. Scale bars: 20 μm.
Discussion
Previous studies have shown that relative or absolute podocyte depletion is the key to the initiation and progression of lesions seen in FSGS [18–20]. The most compelling evidence to date for the central role of podocytes and their slit diaphragms in the development of FSGS is the identification of genes mutated in human hereditary FSGS [1]. The products of these genes (nephrin, NEPH1, podocin, CD2AP and α-actinin-4), without exception, are localized to podocytes and their slit diaphragms, and most of them participate in signaling events that are essential for maintaining the podocyte architecture and the slit diaphragm [1, 21].
In the present study, we identified a novel regulator of MP cytoskeletal organization that may be implicated in FSGS. First, we demonstrated the expression and localization of FAM40A in MPs. Secondly, our findings indicated that FAM40A was localized in the nucleus and perinuclear zone. Moreover, based on bioinformatic analysis of FAM40A, it was predicted to have three transmembrane domains binding to DNA, which possibly makes it a membrane protein in the nuclear membrane.
Next, we observed the role of FAM40A in regulating cytoskeletal dynamics and cell morphology of MPs. We successfully constructed three podocyte models (FAM40A-RNAi, FAM40A-WT and FAM40A-MU over-expression) to observe changes in the expression of cytoskeletal protein F-actin. It was found that in differentiated MPs, the actin cytoskeleton was rearranged into fibroblast-like stress fibers extending into foot processes, and F-actin was the dominant microfilament [22, 23]. In FAM40A-RNAi differentiated podocytes, F-actin was increased around the cell periphery and diminished in the cytoplasm. These findings are consistent with those of previous studies, which showed that FAM40A depletion increased the level of cortical actin filaments and often altered the membrane morphology in HeLa cells [6]. Conversely, when the expression of FAM40A increased, the sharp podocytes turned into bigger and round ones with irregular edges, and the structure of primary foot processes was destroyed. The distribution of F-actin was localized to podocyte edges. In FAM40A-mutant differentiated podocytes, sharp cells underwent obvious morphological changes, and F-actin was more marginalized, with decreased expression of nephrin. Furthermore, when the expression of FAM40A varied, the expression of nephrin in podocytes was decreased. These results suggest that FAM40A is a new regulator of podocyte cytoskeletal organization that causes F-actin reorganization and affects the expression of nephrin in the slit diaphragm.
Interestingly, FAM40A is conserved from fungi to human, although we are unable to find a homologue in plants [6]. In human cells, FAM40A has recently been isolated as part of a striatin-interacting phosphatase and kinase (STRIPAK) complex, which also contains PP2A phosphatases, striatins, MOB1/3 and members of the Ste20 kinase family [7]. Therefore, FAM40A is also called STRIP1 (striatin interacting protein 1). Although the functions of STRIPAK complexes remain to be determined, they possibly play roles in signaling, cell cycle control, apoptosis, vesicular trafficking, etc. In addition, STRIPAK complexes were found to be associated with cardiac disease, diabetes, autism, and cerebral cavernous malformation [6, 24]. As a core component, FAM40A may play an important role in STRIPAK complexes. Interestingly, the latest studies showed that FAM40A affected the arrangement of F-actin in tumor cells [6]. Further literature reports suggest that the possible mechanism may be associated with the ability of FAM40A to negatively regulate MST3 and 4 by enabling the formation of a complex containing MST3 and 4 and PP2CA and B. MST3 and 4 are linked to the localized regulation of pMLC and pERM [17]. These studies reveal that FAM40A is closely related to the dynamic regulation of the cytoskeleton.
We confirmed that FAM40A was involved in the cytoskeleton of renal podocytes, which further affected cell morphology, foot processes, and the expression of nephrin in the slit diaphragm. With respect to its effect on foot processes, we noted that both FAM40A and PP2A were the core members of STRIPAK complexes, and that PP2A was a serine/threonine kinase that could induce a variety of physiological changes [25, 26]. In glomerular podocytes, the expression of PP2A only appeared in podocyte foot process formation [27]. Okada acid specificity inhibited foot process formation by acting on PP2A, which affected the formation of primary foot processes and the distribution of F-actin [28, 29]. Both FAM40a and PP2A belong to STRIPAK complexes. The effect of FAM40a on podocyte foot processes may be related to PP2A.
As a functional protein of the slit diaphragm, nephrin plays an important role in maintaining the function of the glomerular filtration barrier. Decreased expression of nephrin could lead to apoptosis, shedding and fusion of podocytes [30, 31]. Here we found that FAM40A leads to decreased expression of nephrin, and it may contribute to clinical proteinuria and nephropathy.
In conclusion, FAM40A participates in signaling events by dynamic regulation of the podocyte cytoskeleton, which is essential for the maintenance of podocyte cytoarchitecture and the slit diaphragm. FAM40A mutation may change the normal cytoarchitecture of podocytes and the slit diaphragm, thus affecting the function of the glomerular filtration barrier. It may result in clinical proteinuria and give insights into the pathogenesis of FSGS. Although many genes have been identified as mutant in hereditary FSGS over the last two decades, no study has addressed pathogenic genes in Chinese cohorts of ethnic FSGS. Our findings may shed light on the role of FAM40A in FSGS and help understand the pathogenesis of FSGS, especially in Chinese populations.