Introduction
Glycogen is a complex/polymeric physiological glucose carbohydrate found mainly in the liver but also in skeletal muscles, kidneys and enterocytes. Its role is to store glucose, which is used as an endogenous source of energy following enzymatic digestion (glycogenolysis process) within 3–4 h after the meal in order to maintain normoglycemia. Glycogen is, therefore, a source of glucose obtained from hepatocytes during a short period of fasting or used for the needs of skeletal muscles in the first early phase of myocytes [1]. Glucose deficiency in the central nervous system (CNS) can lead to neuroglycopenia [2]. There might be symptoms similar to hypoxia, convulsions, irritability and cognitive decline. Permanent and prolonged hypoglycemia may lead to permanent CNS damage [3]. Systemic hypoglycemia is characterized by signs of sympathetic stimulation/activation, including tachycardia, excessive sweating, fatigue, muscle weakness, irritability, anger, hunger, seizures, headaches and dizziness [4].
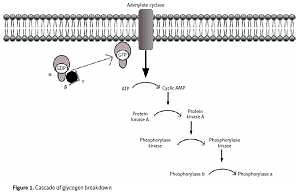
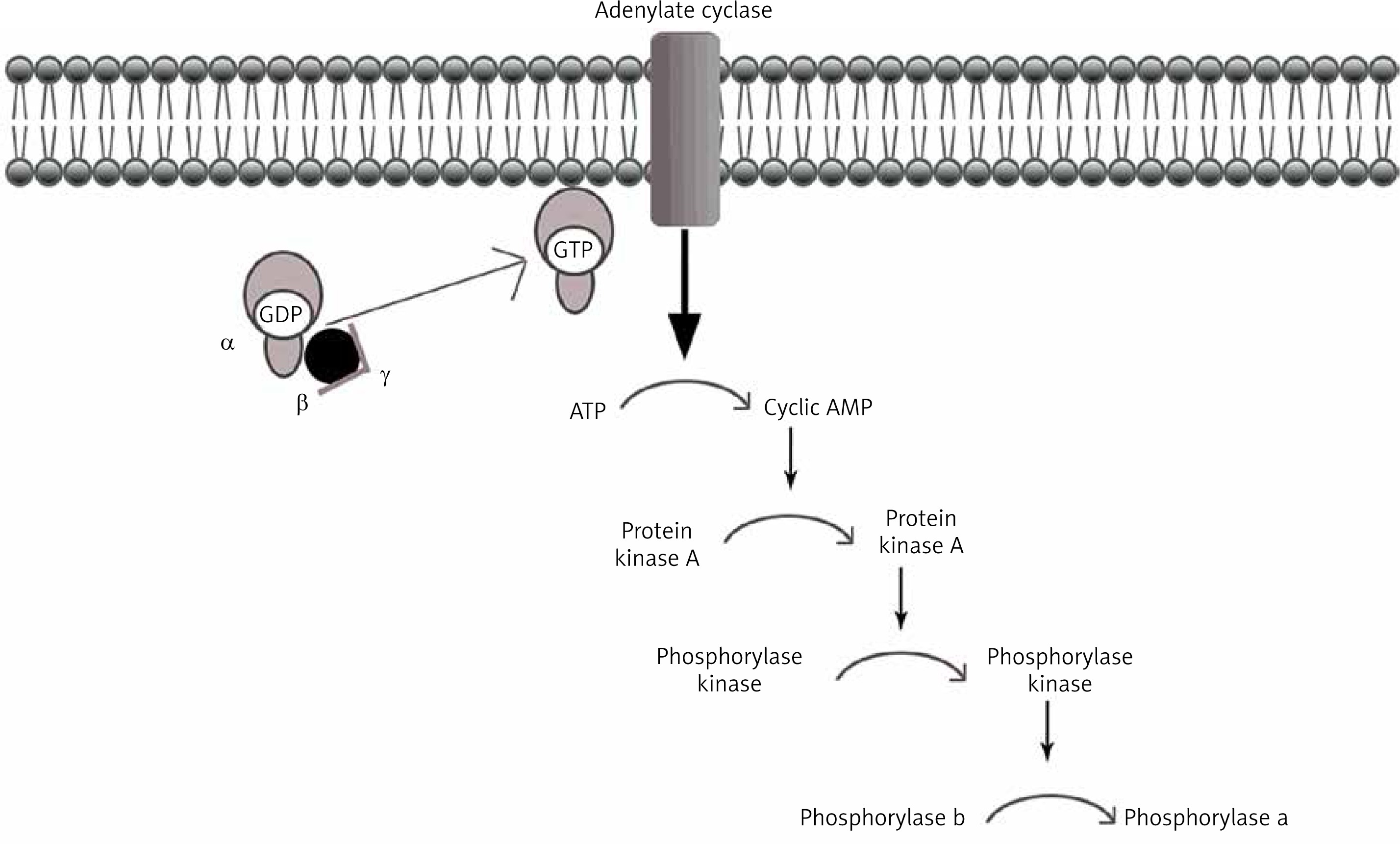
Under physiological conditions, the liver plays a key role in the metabolism of consumed carbohydrates by storing them in the form of glycogen, thereby preventing postprandial hyperglycemia [5]. Synthesis of glycogen consists in combining glucose monomers with glycogen protein and α1,4- or α1,6-glycosidic bonds [6]. This process can be quickly reversed when the energy requirements in the body cells rise again, and the blood glucose levels fall (Figure 1).
Glycogen itself is a branched glucose polymer that is osmotically passive [7]. Due to the activity of many enzymes, hormone regulation and proper conditions in the cell, a healthy body “regulates” and “adjusts” glucose metabolism processes, sometimes “giving warning signals” when it is no longer able to use its own energy resources [3]. Irregularities in the above-mentioned metabolic processes cause problems for the basic functioning of the body [8]. Changes related to the disorder of glycogen metabolism are characterized by symptoms varying from developmental delay, especially of motor functions, to serious and immediate neuroglycopathy, which is a life-threatening condition. In addition, as a result of the accumulation of abnormal glycogen molecules, fatty liver occurs, and in extreme cases the storage of abnormal glycogen forms lead to fibrosis and cirrhosis (GSD type IV) [9].
To keep blood glucose levels within a narrow range (70–140 mg/dl) around the clock, hepatic absorption of postprandial glucose is counterbalanced by its “peripheral” (through other organs) consumption. When the blood glucose level drops below the critical lower limit (long after food intake or effort), the body is ready for a certain compensation and attempts to derive it from non-sugar precursors (for example alanine), but some processes cannot be replaced or stopped [10]. An example is metabolic acidosis with increased blood lactate level with no characteristic symptoms. It is associated with accumulation of lactic acid in cells above 5 mmol/l [11]. This condition results from acid-base balance disorders, and the symptoms are weakness, nausea, vomiting, diarrhea, and abdominal pain [11]. The glucose metabolism pathway in the body can be modulated by using, among other things, a diet rich in complex carbohydrates [12].
In this manuscript, the authors discuss GSDs in general in the first part of the paper, and then, later in the text, they focus only on the hepatic types.
Glycogen storage diseases (glycogenoses)
Pathophysiology and epidemiology
Glycogen storage disease (GSD) is caused by a genetically determined metabolic block involving enzymes that regulate synthesis (glycogenesis) or glycogen breakdown (glycogenolysis) [13]. The nature of the individual types of glycogenosis is related to the impaired accumulation of abnormal molecules of this branched polysaccharide [14]. Moreover, GSD, although it is not a semantically correct name, also refers to a deficit in glycogen synthase (the so-called type 0 GSD), which consists in the lack of glycogen in tissues due to the inability to polymerize glucose molecules (lack of enzyme catalyzing the glycogen synthesis reaction) [15].
In the International Statistical Classification of Diseases and Related Health Problems (ICD-10), glycogenosis is placed under the symbol E74.0 [icd10.pl].
Most genetically determined diseases are rare [16]. These are chronic conditions, usually progressive and mostly severe. The prevalence of metabolic blocks associated with glycogen is estimated at 1 : 25,000 (40,000) births [17].
The typical, biochemical characteristics of this type of GSD are postprandial hyperglycemia (blood glucose cannot be stored in the liver and therefore it is kept high in the blood) with subsequent hypoglycemia (no hepatic glycogen storage from which the body could ‘boost’ the reserves in case of a drop in the blood glucose).
Individuals affected by glycogenosis in Poland may apply for a disability ruling due to the disorder which, irrespective of the source of its occurrence, causes disorder of the body functionality or limitation in performing life and social activities.
Inheritance
Glycogen storage diseases, like most metabolic diseases, are inherited in an autosomal recessive (AR) way. These are the innate defects of carbohydrate metabolism that affect the liver, muscles or both of these organs (the so-called mixed types) [18, 19]. The degree of consanguinity determines the AR inherited diseases in a given area or population – where the risk of mutation is high, which does not exclude de novo mutations. The IX-alpha glycogenosis is the only GSD type which is X-linked and inherited recessively [20].
Typology of GSDs
It is still an unsettled matter. There are categorizations according to different evaluation criteria: author [6], enzyme [21], occupied tissues [17] or mutation location. The classification by author in the order of discovering/describing individual GSDs is generally accepted and applied. The nomenclature comes from the author of the pioneering study, the method of treatment, or the diagnosis of the case of glycogen storage, since the time when this disorder was not classified as a separate disease unit. This division can also be found in all sorts of general papers on medical biochemistry, genetics, and in pediatrics or neonatology.
Another attempt to classify GSDs is the Shin [21] study, where the glycogenosis classification is precisely categorized according to the enzymatic substrate of glycogen metabolism and the subtypes associated with certain organs, giving a total of 12 GSDs (Table I).
Table I
Various types of GSDs (types of GSDs according to tissue-specific enzymatic deficiency)
Özen [17] classifies GSDs on the basis of the latest knowledge in the field, their enzymatic deficits or involved tissues, mainly on the basis of available case studies. Case studies and data collection during patient health monitoring prove an increase in the number of GSDs studies [18, 22, 23]. The knowledge and awareness of these complex metabolic pathologies are increasing. The author divided the aforementioned disorders into hepatic, mixed, and muscular GSDs (Table II) [17].
Table II
Classification of GSDs into hepatic and muscle types with separate classification of PhK deficiency (type IX)
Clinical symptoms
The basic clinical symptoms common for every type of hepatic GSD are hypoglycemia and hepatomegaly [24]. A characteristic feature for muscle GSDs is progressive muscle pathology, including exercise-induced muscle weakness [25]. Metabolic acidosis with hypercholesterolemia and hyperlipidemia are found in blood biochemistry [26]. In both types of GSDs, there is also hypertransaminasemia (elevated activity of liver enzymes – aminotransferases). Physical development delay, mostly in the form of short stature or motor latency, is a typical clinical symptom. GSDs are a group of heterogeneous genetic diseases; therefore each type has its distinct, specific clinical presentation [27].
Diagnosis
The diagnosis of GSD is based on clinical signs and/or symptoms related to hypoglycemia and hepatomegaly. Laboratory parameters such as increased lactate level (in GSD type I), elevated level of serum cholesterol and triglycerides and hypertransaminasemia are helpful in diagnosis establishment. The key point is to differentiate between GSD type I and so-called ketotic types (III/VI/IX). Symptoms in GSD I typically present earlier (in the first few months of life) with severe fasting hypoglycemia within 3–4 h after feeding. Hypoglycemia is usually less severe in patients with GSD III/VI/IX due to the intact process of gluconeogenesis [28]. Blood lactate levels increase rapidly in GSD I as blood glucose (BG) concentrations decrease to levels that normally trigger a counter-regulatory response (< 70 mg/dl or 4 mmol/l) and are markedly increased when BG levels decrease to < 40–50 mg/dl or 2.2–2.8 mmol/l). Blood β-hydroxybutyrate levels increase only modestly in GSD I, in contrast to marked hyperketonemia with fasting hypoglycemia characteristic of GSD 0, III, VI, and IX [29].
Other biochemical characteristics that help to distinguish between these disorders are elevated uric acid and lactate levels in GSD I, whereas these are typically normal in ketotic GSDs [30].
The final diagnosis is always established or confirmed with molecular tests. Liver biopsy, in order to assess enzymatic activity in hepatocytes, is no longer the gold standard – it has been replaced by genetic examination, which is non-invasive and gives a certain and final diagnosis [29, 30]. The disadvantages of molecular testing are its still high costs, limited availability and long time of waiting for the results.
The Children’s Memorial Health Institute in Warsaw (Department of Pediatrics, Nutrition and Metabolic Diseases) is the reference center for metabolic diseases.
Glycogen pathway
Decomposition of glycogen molecules is directly related to energy production, and its regulation takes place by activation of the relevant substances and enzymes. In a healthy body, glycogen metabolism is effective through the balance between glycogen synthase and phosphorylase activity (Figure 2). Both enzymes can be reversibly phosphorylated in more than one site by separate kinases [8].
Glycogenolysis (branched glucose polymer phosphorolysis) is carried out in three steps:
Release of glucose-1-phosphate-kinase phosphorylase (PhK).
Conversion of glucose-1-phosphate to glucose-6-phosphate – phosphoglucomutase.
Glucose-6-phosphate metabolism – glucose 6-phosphatase-6-phosphatase 1 (G6PC).
Glycogenolysis takes place in the liver, during hunger or in the muscles during intensive exercise, to compensate for a decrease in blood glucose levels or a lack of chemical energy carriers in cells – ATP [31].
In the early phase of glycogenolysis, the released glucose is already phosphorylated and can undergo further transformation. In this state it cannot freely leave the cell, which makes it a very energetically beneficial reaction for the working muscle. However, there are circumstances in which glycogen metabolism is inadequate.
GSD type IX
The GSD type IX accounts for about 25% of all glycogenosis and consists of a lack of phosphorylase kinase (PhK) enzyme, which plays a role in the process of glycogen degradation. The enzyme occurs in many tissues, thus its subunits also have their specific isoforms. The activity of PhK has been studied in such organs as the liver, muscles, kidney, testes, heart and also in erythrocytes, leukocytes and nerve cells [32, 33].
The PhK enzyme consists of 4 subunits: α, β, γ, and δ subunits, which are encoded by the following genes:
The α and β subunits occupy 4/5 of the total weight of the holoenzyme and have a superior function that regulates phosphorylation. The cAMP-dependent protein kinase regulates the phosphorylation of the Ser residues in the α and β subunits. The γ subunit contains a catalytic site, and the calcium-binding subunit δ has an affinity for calmodulin [36].
Type IX is the only GSD that is X-chromosome linked recessively inherited: the α1 subunit is X-linked inherited, while the remaining subunit units are AR inherited [37]. Mutation in the PHKA2 gene is the most common cause of GSD IX [38]. This type of GSD is characterized by a relatively mild course. Tissues affected by the disorder, depending on the subtype, are liver, erythrocytes, kidneys and muscles [39, 40]. The symptoms occur during infancy or early childhood, and include growth delay and liver enlargement [41]. Episodes of hypoglycemia or ketonuria without a reasonable cause are rare. If present, they are associated with a prolonged state of starvation or increased physical activity. Lactic acid and urinary acid concentrations are usually normal, and metabolic acidosis with hypocalcemia is rare. Hypertriglyceridemia and hypercholesterolemia, as well as various levels of hypertransaminases, are also observed: from slightly to several times elevated transaminases. It causes delays in motor development due to the possibility of spreading in the muscle tissue. These disorders normalize during adolescence [13]. Occasionally, these GSDs are associated with more serious complications such as cirrhosis of the subtype IX γ or cardiomyopathy/megalocardial dysfunction [42–44].
GSD type III and VI
Type III of GSD is caused by a reduced enzyme degradation enzyme (amylo-1,6 glucosidase + transferase – GDE), which is a key enzyme in the glycogenolysis process [45]. Based on the additional involvement of the skeletal muscles and the heart, there are 2 subtypes of GSD III: hepatic-muscle type (subtype a) and a hepatic one (subtype b). Like all other GSDs but type IX-α, it is inherited in an autosomal recessive manner; the mutation is in the AGL gene located on chromosome 1p21 [46].
The first signs of the disease appear in early childhood and include enlarged liver, delayed growth and physical development, whereas hypoglycemia (neurohyglycemia) is not that common. As age progresses, the hepatomegaly reverses, and muscle weakness (subtype IIIa) progresses slowly. Other symptoms common to this type of GSD include muscle hypotension and cardiomyopathy. Frequently, the symptoms regress during adolescence, except in rare cases when cirrhosis of the liver or myopathy occurs [47].
Type VI is a mild form of GSD. The enzyme block consists of decreased liver enzyme activity – phosphorylase involved in the glycogenolysis process. The disease is inherited in an autosomal recessive manner, with a mutation in the PYGL gene on chromosome 14q21-q22 [48]. Symptoms are the same as in type III. In this type of GSD the heart and skeletal muscles are never involved, and liver adenomas are very rare. The disease does not carry the risk of organ failure [49].
GSD type IV
Type IV is a rare and severe form of GSD, accounting for approximately 3% of all types of glycogenosis. The enzyme defect consists of reduced glycogen brancher enzyme (GBE) activity, mutation in the GBE1 gene on chromosome 3p12, and autosomal recessive inheritance. GBE deficiency results in the accumulation of abnormal forms of glycogen that resemble the amylopectin (polyglucosan body) structure. Hence, its other name is amylopectin or adult polyglucosan body disease (APBD) [50].
The symptoms are very heterogeneous and include both the liver and the neuromuscular system. Children are generally born healthy but, as early as in the first months of life, they develop hepatomegaly and hypotonia and their psycho-motor development is delayed. The disease progresses rapidly. It leads to liver fibrosis and portal hypertension which manifests as ascites and eventually leads to death. However, several cases of non-available hepatic GSD IV have also been reported in the literature [51].
GSD type I
Unlike muscles, liver contains the glucose-6-phosphatase membrane enzyme, which removes the phosphate residue to allow the glucose to enter the bloodstream and regulates its concentration [52].
Glucose-6-phosphatase 1 (G6PC) enzyme catalyzes the hydrolysis of glucose-6-phosphate to glucose, thus forming the last step of glycogenolysis and gluconeogenesis [53–56]. The G6PC enzyme is encoded by the G6pc gene, which is expressed in the liver, kidneys and pancreas, and its mutation is inherited in an AR pattern [52, 57]. The function of the enzyme requires its translocation through the membrane of the endoplasmic reticulum. Another enzyme, G6PC translocase encoded by the SLC37A4 gene, is also involved in this process, which can also be mutated (AR inheritance) to impair the function of neutrophils, which is inscribed in the GSD type Ib.
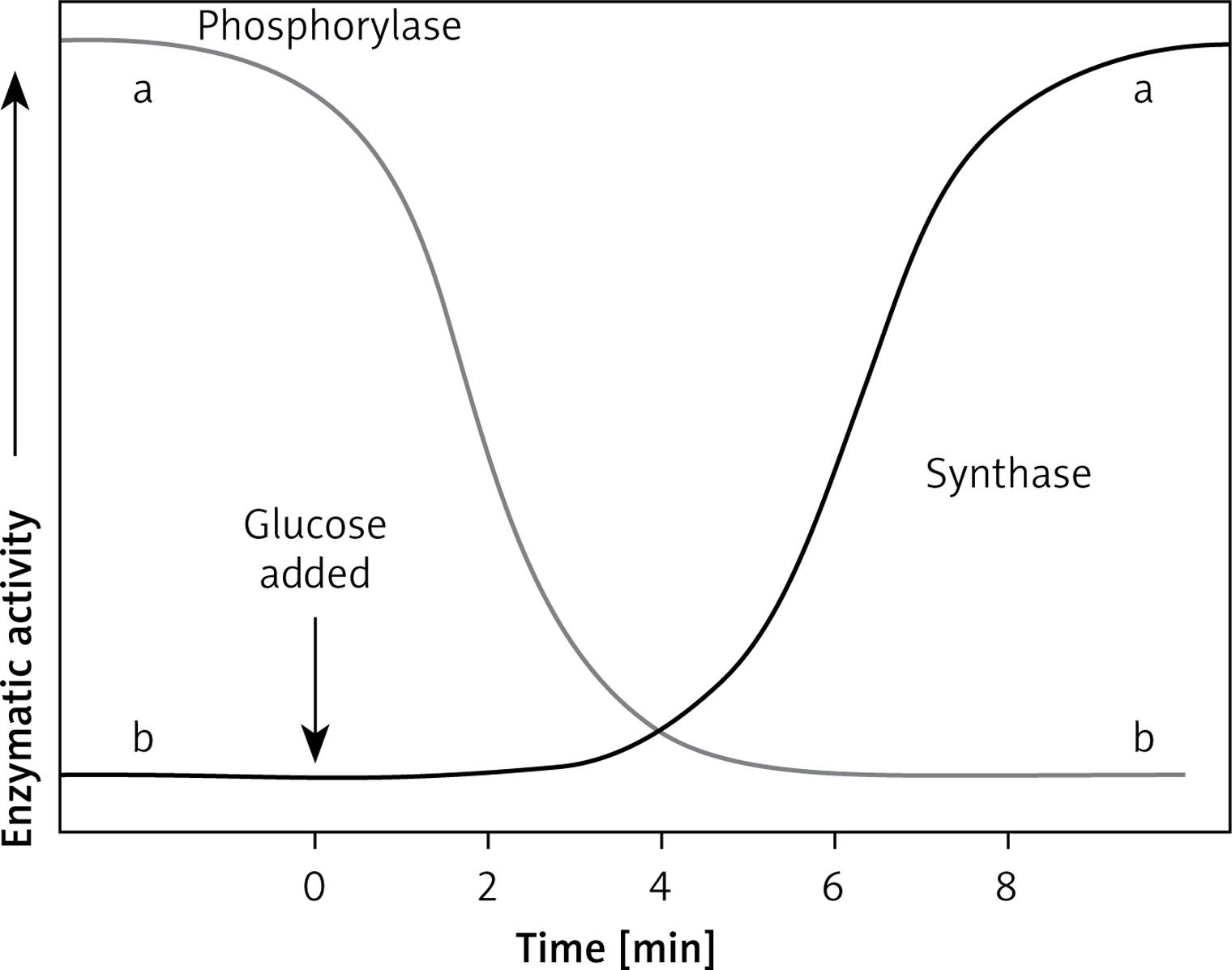
After eating a meal, blood glucose rises. Hormone levels regulating these metabolic pathways are in inverse proportions, i.e. the concentration of glucagon decreases, and the concentration of insulin increases. There is no breakdown of glycogen then. The phosphorylation pathway of inactivated phosphorylase (b) to active (a) is off. On the other hand, the pathway of phosphorylation of active synthase (a) to inactive (b) is activated. We are dealing with the postprandial state of glycogen synthesis stored as a backup material in tissues, primarily in the liver. Before meals, when blood glucose levels fall, insulin levels drop, and another hormone, glucagon (adrenalin-like), shows an upward trend. The glycogen degradation reaction to glucose is started. We obtain an active form of glycogen phosphorylase (a) and an inactive form of glycogen synthase (b).
Dietary management
The basic treatment for GSDs is dietary management, which raises many controversies. Dietary recommendations differ slightly between the United States and Poland, but they are different in Europe, for example, comparing the UK and Poland.
The aim of dietary treatment is to avoid hypoglycemia. Therefore, it is necessary for patients to consume frequent meals during the day (every 3–4 h) with the addition of raw cornstarch (RS) and to shorten the night break by an additional portion of RS in the middle of the night [58–60]. Starch, like glycogen, is a polysaccharide – the process of releasing pure glucose into the bloodstream, in this case, is extended in time. This results in constant access of substrates to the biochemical reactions of energy synthesis pathways, limiting its accumulation in tissues and thereby involving glycogen [61, 62]. In the UK, overnight glucose infusions through a pump or probe are still used.
Studies have confirmed that a snack before bed time is a sufficient energy/glucose supply for patients with GSD type III/VI/IX. However, for patients with an unstable glucose level or ketosis, 2 g of unprocessed RS per kilogram of body weight is prepared to prevent morning hypoglycemia [63]. The situation is different for patients with type I GSD, who present with the most severe hypoglycemia and have most unstable glucose levels – both glycogenolysis and gluconeogenesis are impaired in this type and, additionally, ketogenesis is ineffective and there is a risk of neuroglycopenia. Therefore, in GSD type I starch is a drug and not just a dietary supplement; it must be given regularly during the day every 3–4 h with a night break not exceeding 7–8 h [30]. Patients most often need to take an extra dose of starch at night while maintaining a constant supply of carbohydrates in a liquid form, especially during sleep, thus protecting patients with decompensated glycemia from dangerous complications of hypoglycemia.
When intolerance or reluctance to receive RS is observed, either a nasogastric tube or percutaneous endoscopic gastrostomy (PEG) is required – if the enteral supply is to be maintained for more than 6–8 weeks (ESPEGHAN guidelines). The disadvantage of starch is that it is hard to digest – with an infection associated with gastrointestinal irritation in children with glycogenosis there are often problems with starch supplementation. In addition to child monitoring, an indispensable element in hypoglycemia and RS treatment is blood glucose measurement with a glucometer, and in the case of GSD III/VI/IX ketones in the urine are to be measured. In these types and in type Ia, it is possible to provide a modified, artificially produced starch called “Glycosade”, which, due to its prolonged release, allows normoglycemia to be maintained for an 8-hour nighttime rest. It is not recommended for patients with type Ib due to their common inflammatory bowel disease and, therefore, additionally impaired absorption. The formula is used in patients older than 5 years old. Currently, glycosade is to be studied in the United States for its application during the day in adult patients, in whom, due to the slower metabolism than children, the extended-release preparation might be effective not only at night. According to current US dietary recommendations, the diet should be restricted to simple sugars (less than 5–10 g in each meal) in all types of GSDs, while it is most restrictive in type I, with RS supplementation (in type I every 3–4 h daily plus an extra overnight dose or only 1 night dose in other types) [64].
Type III/VI/IX is recommended to use the so-called high-protein diet (GSD type III; 3–4 g/kg BW, VI and IX: 2–3 g protein per kilogram BW per day) [65]. Because of the increased risk of micronutrient deficiencies (vitamin D3, calcium-phosphate disturbances, and the risk of osteopenia), multivitamins and vitamin D3 supplements are also recommended [30, 64]. The basic principle of treatment is therefore to limit the simple sugars in a diet rich in complex carbohydrates. However, the diet is always individually selected for the patient based on its glycemic status, current biochemical findings (metabolic equilibrium) and anthropometric parameters, and requires close collaboration between the physician, metabolic dietitian, the patient and his family.
Table III summarizes different dietary strategies in ketotic and non-ketotic (type I) GSDs.
Table III
Summary of different dietary strategies in ketotic and non-ketotic (type I) GSDs
| Dietary management | |
|---|---|
| All GSDs | |
| GSD I: | GSD III/VI/IX: |
As mentioned above, a properly applied specific diet is a treatment in GSDs. It leads not only to stable normoglycemia, but also to decreased hepatomegaly (reduction of glycogen storage), improvement of growth and biochemical metabolic control (normalization of transaminases, triglycerides, in type I – reduction of lactate and uric acid in blood) [13].
The problem of diet, as an important factor in treatment, may lead to eating disorders in children with GSDs. In the course of metabolic diseases, some eating disorders related strictly to the (elimination) diet can be observed. For example, people consuming frequent meals with high carbohydrate content that slows down the release of glucose into the blood may experience a lack of hunger. Since a lot of attention is paid to eating, it may cause a lack of pleasure from eating a meal, not to mention the lack of taste of non-sweet foods. As far as children are concerned, following the heavy-starch diet is often associated with digestive problems. Children who have previously consumed sweet products being deprived of candy may feel alienated among their peers, in a particular environment, for example, in a pre-school class where the group’s diet contains sweet supplements, unlike meals prepared for a child with GSDs. Establishing the right diagnosis and setting a proper glycogenic diet will make the patients enjoy eating and discovering new flavors, changing the attitude to a meal as a necessity. In the treatment of eating disorders, therapies of these disorders adapted to the requirements of GSDs are helpful [28].
The GSDs, like all genetic diseases, is incurable. Currently, clinical trials on gene therapy for type I glycogenosis (clinical phase of GSDI adult safety assessment) are ongoing at the Connecticut facility in the US [66].
Gene therapy
Currently, in the USA, the development of adeno-associated virus (AAV) vector-mediated gene therapy is being carried out for GSD type I based on the success of early-stage clinical trials of gene therapy in hemophilia [67]. So far, AAV vectors containing a human G6Pase regulatory cassette/promoter have proven to be efficacious in animal models of GSD Ia, and these vectors contain sequence elements that regulate G6Pase expression appropriately [68]. At present, gene therapy for GSD type I is at the stage of a safety clinical trial on adult patients with this type of GSD, and is taking place in the Connecticut Hospital within the GSD program of Prof. Weinstein.
Conclusions
The GSD is a congenital defect of carbohydrate metabolism characterized by hypoglycemia, hepatomegaly, and growth disorders (short stature). Basic therapeutic treatment consists in maintaining a proper diet with RS supplementation.
Due to the fact that awareness and knowledge about rare diseases are still insufficient, it is important to popularize them among pediatricians, hepatologists and geneticists. Knowledge of the biochemical basics and glucose metabolism in the human body facilitates proper treatment of GSD.