Introduction
β-Globin protein (HBB), one of the hemoglobin subunits, is produced by β-globin gene (HBB), which is located on chromosome 11 [1]. Two β-globin molecules bind to two α-globin molecules to constitute the most popular form of hemoglobin, adult hemoglobin (HbA). HBB is crucial in balancing the ratio of (α : β)-globin chains, preventing the aggregation of insoluble α-globin complex [1–3]. Abnormality in the HBB gene results in an inherited recessive blood disorder that can be caused by variants at the transcription or translation level affecting the stability and the production of the β-globin chain [4, 5].
Several HBB variants are produced by mutations in HBB gene, and some mutations alters the production of HBB chain either partially (β+) or completely (β0) [6]. Reduced formation of HBB chain lowers the amount of functional Hb, which is a characteristic of the highly prevalent blood disorder in Saudi Arabia, β-thalassemia [7–12]. Variations in HBB protein can also be associated with other genetic hematological disorders such as sickle cell disease, which is very common in Saudi Arabia. To date, there are more than 1,700 hemoglobin variants that have been reported, with more than 900 variants in HBB [13]; however, the most common variants are hemoglobin E (HbE), sickle hemoglobin (HbS), and hemoglobin C (HbC) that results as a consequence of point mutations in HBB gene [14]. The formation of the highly unstable α and β complex HbE is a result of a point mutation at position 26 in HBB that substituted glutamic acid with lysine, causing a phenotypic characteristic of a mild form of β-thalassemia [15]. During the production of HbS, the most common cause of sickle cell disease is a consequence of a point mutation at the sixth position in HBB gene that substituted glutamic acid codon (GAG) with valine codon (GTG) [16]. Additionally, glutamic acid can be substituted with lysine forming HbC, which is a Hb variant that is related to sickle cell disease [5]. On the other hand, some variants might have beneficial influences. For instance, it has been known that individuals with HbC are protected at different level against Plasmodium falciparum that cause malaria [17, 18].
The severity of the disease relies on some factors such as the variant’s characteristics and the imbalance ratio of α : β chains [19]. There are about 42 HBB variants that have been identified in the population of Saudi Arabia [7]. These mutations either reduce (β+) or prevent (β0) the synthesis of HBB protein. Therefore, identification of HBB genes variants will be helpful to understand the heterogeneity and prevalence of pathogenic variants in the population of Eastern Province. Thus, this study was conducted in an effort to identify the HBB gene variants in the population of the Eastern Province of Saudi Arabia.
Material and methods
Samples collection
A total of 545 blood samples were collected from male (n = 303) and female (n = 242) participants from the Eastern region of Saudi Arabia. This study included 215 transfusion-dependent subjects (age range: 2 months to 51 years; 106 males and 109 females) and 330 normal healthy subjects (age range: 8 months to 67 years; 197 males and 133 females), who attended major hospitals in the region. Samples were requested from random volunteers and patients who were clinically diagnosed with β/α-thalassemia major/carriers or sickle cell anemia. Transfusion-dependent subjects include 20 sickle cell disease (homozygous) patients and 195 β-thalassemia major patients. Subjects with no history of blood transfusion, blood disorders, or chronic diseases were included as normal control.
The study was approved by the Standing Committee for Research Ethics on Living Creatures, Imam Abdulrahman Bin Faisal University (CBME2012032; IRB-2015-08-069).
Hematological parameters
Hematological parameters were evaluated following the collection of blood samples (5 ml) in EDTA-coated vacutainers using Coulter Micro Diff II (Beckman Coulter, Inc., Brea, CA, USA) and VARIANTTM II Hemoglobin Testing System (BIO-Rad Laboratories, Inc., Hercules, CA, USA).
DNA extraction and sequencing
DNA was extracted from all of the 545 blood samples utilizing QIAamp DNA blood minikit (Qiagen, GmbH, Hilden, Germany) followed by amplification of HBB, HBA1 and HBA2 genes using the PCR standard method [8, 20–22]. Utilizing forward and reverse primers separately, HBB, HBA1, and HBA2 genes in each sample were amplified by PCR using BigDye Terminator Cycle Sequencing Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Following the amplification of each gene, purification was conducted to prepare the samples for sequencing, which was performed using the series Genetic Analyzer 3500 (Thermo Fisher Scientific, Inc.) at the Department of Genetic Research, Institute for Research and Medical Consultation, Imam Abdulrahman Bin Faisal University (Dammam, Saudi Arabia). DNA sequencing analysis software v. 5.3 (Applied Biosystem; Thermo Fisher Scientific, Inc.) and mutation surveyor software (Softgenetics, US) were used to analyze electropherograms.
Results
Conducting a direct sequence of HBB gene from 545 (215 transfusion-dependent subjects and 330 normal healthy subjects) subjects revealed that the 249 subjects were with normal HBB gene and 296 subjects were identified with 36 (HBB:c.118C>T; HBB:c.315+1G>A; HBB: c.93-21_96del; HBB:c.20A>T; HBB:c.92+5G>C; HBB:c.92+6T>C; HBB:c.17-18delCT; HBB:c.93-21G>A; HBB:c.25_26delAA; HBB:c.9T>C; HBB:c.27_28insG; HBB:c.79G>A; HBB:c.92+1G>A; HBB:c.431T>A; HBB:c.2T>C; HBB:c.112delT; HBB:c.315+74T>G; HBB:c.415G>A; HBB:c.432C>T; HBB:c.435G>A; HBB:c.281G>T; HBB:c.26A>T; HBB:c.92+2T>C; HBB:c.218G>C; HBB:c.253A>G; HBB:c.294C>T; HBB:c.135delC; HBB:c.93-1G>C; HBB:c.370A>C; HBB:c.320T>C; HBB:c.315+208T>G; HBB:c.315+241T>A; HBB:c.315+376T>C; HBB:c.316-183_316-168del; HBB:c.252C>T, and HBB:c.316-114C>G) different types (β0: a complete lack of β-globin production; β+: a partial production of β-globin; βS: sickled; βi: intronic mutation; whereas βE is HbE disease) of variants in the exonic and intronic regions of HBB gene. Among the 36 HBB variants, 25 variants have been previously reported from the Saudi Arabian population, the remaining 11 variants have been identified for the first time in this study in Saudi Arabians. Among these 11 variants, 7 (2 exonic and 5 intronic) variants have been identified for the first time in HBB gene in the study subjects. These 7 variants (HBB:c.281G>T; HBB:c.316-183_316-168del; HBB:c.315+208T>G; HBB:c.315+241T>A; HBB:c.315+376T>C; HBB:c.252C>T; and HBB:c.316-114C>G) have not been reported earlier in any population; hence, these 7 variants were described in detail in Table I and are referred as novel variants.
Table I
Hematological features and genetic results of subjects with newly identified HBB mutations
Twenty-six HBB variants (HBB:c.118C>T; HBB:c.20A>T; HBB:c.315+1G>A; HBB:c.93-21_96del; HBB:c.92+5G>C; HBB:c.93-21G>A; HBB:c.17-18delCT; HBB:c.25_26delAA; HBB:c.27_28insG; HBB:c.79G>A; HBB:c.431T>A; HBB:c.2T>C; HBB:c.112delT; HBB:c.315+74T>G; HBB:c.415G>A; HBB:c.432C>T; HBB:c.435G>A; HBB:c.281G>T; HBB:c.9T>C; HBB:c.92+2T>C; BB:c.218G>C; HBB:c.370A>C; HBB:c.315+208T>G; HBB:c.315+241T>A; HBB:c.315+376T>C; and HBB:c.316-114C>G) have been observed in 86 subjects included as normal control group. Out of the 86 subjects, 42 were observed with βS, 3 were observed with βE, 22 were observed with β0, 3 were observed with β+, and 4 were observed with βi. Twenty-two HBB variants (HBB:c.118C>T; HBB:c.20A>T; HBB:c.315+1G>A; HBB:c.93-21_96del; HBB:c.92+5G>C; HBB:c.92+6T>C; HBB:c.93-21G>; HBB:c.17-18delCT; HBB:c.25_26delAA; HBB:c.27_28insG; HBB:c.112delT; HBB:c.415G>A; HBB:c.281G>T; HBB:c.316-183_316-168del; HBB:c.92+1G>A; HBB:c.26A>T; HBB:c.253A>G; HBB:c.294C>T; HBB:c.135delC; HBB:c.93-1G>C; HBB:c.320T>C; and HBB:c.252C>T) have been observed in the transfusion-dependent subjects. Where 20 subjects were observed with homozygous βS, remaining transfusion-dependent subjects were observed with homozygous β0, homozygous β+, or with compound heterozygous β0 and β+.
Two males and 3 females transfusion-dependent subjects had a total of four novel variants; 3 point mutations; HBB:c.252C>T, HBB:c.281G>T, and HBB:c.316-114C>G as well as one deletion; HBB: c.316-183_316-168del (Figure 1, Table I). The HBB:c.252C>T is a silent point mutation, where the change in the codon results on encoding for the same amino acid, glycine. The point mutation; HBB:c.281G>T is a missense mutation, which results on encoding for the amino acid phenylalanine rather than cysteine. The point mutation, HBB:c.316-114C>G is present in the intronic region of HBB gene. The newly reported deletion (HBB:c.316-183_316-168del) was observed in two transfusion-dependent patients who are clinically diagnosed with sickle cell anemia. The most common α-thalassemia mutation amongst these transfusion-dependent subjects was the 3.7 single gene deletion. However, one of the subjects had HBA1 intronic mutation (Table I).
Figure 1
Schematic representation of HBB gene structure and novel identified variant locations. The position of the mutations is indicated below the gene. Novel mutations detected in subjects with transfusion-dependent are in red, mutations detected in normal subjects are in green, and the mutation identified in both subjects is in gray
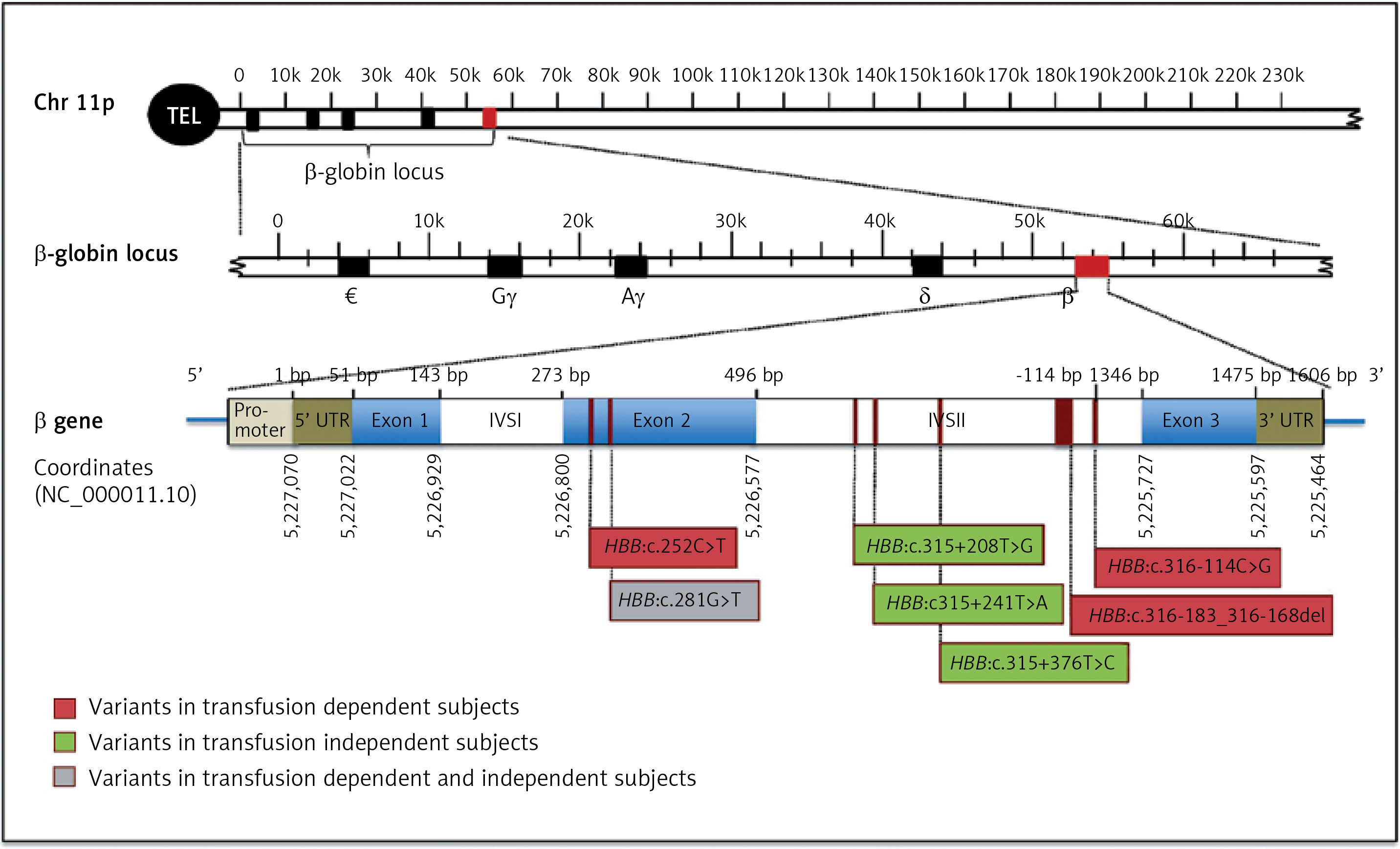
On the other hand, four newly identified point mutations have been observed in normal control participants (2 males, 1 female): HBB:c.281G>T, HBB:c.315+208T>G, HBB:c.315+241T>A, and HBB:c.315+376T>C (Figure 1, Table I). HBB:c.281G>T is a point mutation that was identified also in a normal control subject, which results on the conversion of an UGU codon into UUU, which encodes the amino acid phenylalanine rather than cysteine. The remaining three point mutations: HBB:c.315+208T>G, HBB:c.315+241T>A, and HBB:c.315+376T>C are variants that present in the non-coding regions of HBB gene (Figure 1). These novel HBB variants in normal healthy group are co-inherited with α-globin deletion.
Furthermore, in an effort to test the notion that the presence of these seven novel identified variants would have a more influence on red blood cell pathology compared to control/sickle cell/thalassemia conditions, the general hematological parameters of blood samples were evaluated. In each sample, the co-inheritance of the novel mutation/mutations at the intronic regions seems to demonstrate no influence on blood parameters compared with existing of these conditions alone. However, one of the normal control subjects had a novel exonic missense mutation: HBB:c.281G>T and borderline HbA2 level, i.e. 3% (Table I).
Discussion
HBB and HBA genes encode the normal adult hemoglobin tetramer (Hb), constituting of four polypeptide chains; 2 α chains and 2 β chains [23]. Mutations such as frameshift, minor deletions, and missense mutations in the HBB gene may result in highly unstable HBB protein [6, 24]. Various novel mutations have been still observed in HBB gene. Recently, Ekwattanakit et al. [25] have identified two frameshift mutations in the third exon that causes a rapid mRNA decay in thalassemia intermedia patients. The novel heterozygous mutation identified in this present study, HBB:c.281G>T in exon 2 is caused by a missense mutation that substituted the sulfur containing amino acid, cysteine into the aromatic amino acid, phenylalanine [26]. This substitution might alter the protein structure or interfere with the heme-binding pocket as it has been shown that sulfur containing amino acids form stronger interactions compared to the aromatic amino acids. This substitution might reduce the stability of the protein function [26]. Further studies on these novel mutations are needed to investigate any alteration in the proper function of HBB protein.
HBB produces an important component in building up hemoglobin complex, which carries on oxygen molecules into the cells. The production of β-globin chain affects the ratio of α and β, and in turns the production of an intact hemoglobin complex. In addition, the imbalance of α/β chains causes the formation of toxic complexes of free α chains aggregation [2, 27]. There are several genetics hematological disorders associated with genetic changes in the HBB. This genetic change might yield unfunctional, structurally abnormal β-chain variants, or affect the proteins’ production [4, 5]. However, it has been previously found that the co-inheritance of β-mutations with α-mutations might reduce the amount of α-globin production, which in turn reduce the imbalance of β/α chain resulting in the amelioration of disease severity [28].
It is worth to note that these novel mutations that have been identified in this study are co-inherited with other mutations, which are present in HBB gene or other genes that might directly interact with HBB. Therefore, it cannot be concluded whether the changes in the hematological parameters are influenced by the novel identified variants or as a result of other factors. To illustrate, the novel hetero-exonic mutation HBB:c.281G>T, which has one amino acid change from cysteine to phenylalanine, has been found solely in a β-thalassemia carrier as well as in a β-thalassemia major with other mutations. The β-thalassemia carrier subject had a borderline HbA2 level 3% (Table I), which could be a consequence of either the mutation or the co-inheritance of α deletion [3, 7] as it has been previously shown to increase HbA2 level [27, 29]. The severity of the phenotype in the β-thalassemia major subject might be affected by the presence of other mutations.
In an effort to understand the implications of these novel identified variants, number of future studies needs to be conducted. HBB gene consists of three exons and based on previous studies, the severity of the disease lies on which exon the mutations are located. For example, based on Cao and Galanello [6], the mutation in phase termination codon stimulates the nonsense-mediated mRNA decay (NMD) process, resulting in low production of normal HBB chain [6, 24]. Since the two exonic novel mutations presented in this study are located in exon 2, further studies are required to investigate whether they have similar effects. Similarly, majority of the identified mutations in this study were present in intronic region, which can alter the gene’s function and expression in different ways, as non-coding regions play a role in transcriptional and translational regulation. Non-coding mutations may also introduce novel splice sites that result in a truncated protein, leading in loss of function [30]. In addition, the effect of homozygosity of these mutations is not distinguished. Thus, future studies could overcome this limitation by investigating the effect of inheriting two mutant alleles of these novel variants on the severity of the phenotype.
In conclusion, HBB molecular scanning for both transfusion-dependent and normal control subjects is an important step for better understanding and accurate diagnosis of hemoglobinopathies. This current study has identified 11 variants that have been reported for the first time in Saudi Arabia. Seven novel (2 exonic and 5 intronic) variants in HBB gene have been observed in the study population. The influence of these novel variants on the phenotypic characteristics and β-globin proper function is currently unknown and thus needs to be examined. The identified β-globin mutations could be used as a genetic to the study region.