

Introduction
Glaucoma is defined as a degenerative disease of the optic nerve manifesting as retinal ganglion cell (RGC) death and progressive visual field loss. Increased intraocular pressure (IOP) is the main factor leading to glaucoma. However, there are patients with normal-tension glaucoma, where optic nerve damage progression does not result from elevated IOP, and patients with ocular hypertension (OHT) where increased IOP does not result in optic nerve damage.
The factors that lead to glaucoma neuropathy independently of IOP include a range of molecular and immunological mechanisms. The rodent models of glaucoma contribute to the research [1, 2].
There are inherited glaucoma models, e.g. DBA/2J mice with gene mutations resulting in a state resembling pigment dispersion syndrome [3-5]. Other glaucomatous models include inducing excitotoxicity and oxidative stress in N-methyl-D-aspartate (NMDA) [6] and tumor necrosis factor α (TNF-α) intravitreal injections and optic nerve crush [7, 8].
The ocular hypertension models lead to IOP elevation via diverse mechanisms, e.g. intracameral injections of polystyrene or magnetic microbeads (pre-trabecular models) [9, 10], episcleral vein cauterization [11], laser photocoagulation [12] or hypertonic saline injection [13]. The normal-tension glaucoma models include immunization with ocular antigens – experimental autoimmune glaucoma model [14].
Immunological basics of glaucomatous neurodegeneration
In spite of the lack of lymphatic drainage and the presence of the blood-retina barrier, the immunological and inflammatory reactions in the eye are omnipresent and complex [8].
The RGC death through autoimmunological mechanisms is considered a part of glaucomatous damage. It is reported that S100B immunization resulted in higher MBL and C3 protein concentrations in the optic nerves and higher interleukin 1b (IL-1b) concentration in the aqueous humor. The increase of NFkB in S100 retinas leads to the conclusion that the NFkB induced RGCs loss is triggered via MBL [14].
The main mechanisms of immunological glaucomatous neuropathy that have been studied are the complement, TNF-α pathways and toll-like receptors (TLR) pathways [15-17].
The elevated activity of the complement pathway in the glaucomatous retinas and optic nerves has been found in both OHT animal models [18-20] and IOP-independent animal models [14, 21]. The classical complement pathway is triggered by dead or damaged cells tagged by the C1 complex, which then triggers the C3 convertase activation, resulting in the cleavage of C3 into C3a (anaphylatoxin) and C3b (opsonin). C3a activates C5 convertase and also results in the cleavage of C5 into C5a (anaphylatoxin, chemotactic factor) and C5b (first part of MAC – membrane attacking complex). Thus, C5 convertase produces, among others, the proteins forming the MAC C5b-C9b, which leads to cell lysis. It has been found that inhibition of C1 preserves dendritic and synaptic architecture in glaucoma degeneration [16]. C3 not only plays a role in C5 activation and MAC formation, but also has a protective response in glaucoma [22].
TNF-α was found in elevated concentrations in vitreous, retina, aqueous humor and optic nerve [23-26] as well as in the plasma [27] in glaucoma subjects. It is a neuroinflammatory modulator produced mainly in microglia secondary to IL-1 expression due to activation of the NF-kB pathway [17]. Elevated concentration of TNF-α receptors is found in tissue injury and infection [28].
TNF-R1 knock-out mice’s retinas presented a decrease in RGC loss in optic nerve crush glaucoma model [26]. RGC death induced by TNFα involves both caspase-dependent and caspase-independent components of the mitochondrial cell death pathway and generation of reactive oxygen species (ROS). It has the ability to induce glial nitric oxide production, induce excitotoxicity and promote endothelin-1 (ET-1) synthesis and secretion [25]. Oligodendrocytes are the glial cells that take part in immunological mechanisms around the blood-retinal barrier, which is formed by desmosomes. Oligodendrocyte loss in experimental glaucoma models precedes the RGC loss. They are susceptible to oxidative stress and glutamate toxicity, which is potentiated by TNF-α. The TNF-α suppression results in prevention of oligodendrocyte and RGC loss [24].
Glial cells’ role in optic nerve metabolism
The glial cells in the axon and the retina belong to various populations [8]. The glial responses in both compartments – the retina and the RGC axon – mediate protection signals as well as degeneration signals. However, studies show that critical processes happen in the optic nerve head (ONH) [15].
Astrocytosis is suggested to be one of the earliest changes seen in inflammatory RGC loss processes [29]. Astrocytes are the cells present abundantly in proximity to RGC axon bundles. Astrocytes play a role in the optic nerve injury response [30]. They repopulate and remodel at the crush site, upregulating ciliary neurotrophic factor (CNTF) production, which contributes to glial scar formation. Stressed RGCs or astroglial cells in ONH release proteins called DAMP that trigger the inflammatory responses. The DAMP family includes, among other members, heat shock proteins (HSPs) and tenascin-C, an activator of TLR-4 [20]. Release of HSPs occurs during cell necrosis, or secretion of HSPs in exosomes. During special types of apoptotic cell death (for example induced by some chemotherapeutics), HSPs can also appear on the extracellular side of the plasma membrane. HSPs and tenascin-C may lead to TLR activation in microglia [17].
Glial fibrillary acidic protein (GFAP) expression and CNTF production are signs of astrogliosis in the retina [8, 31]. Glial fibrillary acidic protein expression surprisingly is not elevated in the optic nerve upon injury [7]. In glaucoma, astrocytes are the primary producer of TNF-α and TNF-R1 in the ONH. Astrocytes also produce or respond to molecules such as NO, IL-6-type cytokines and endothelins. The ET-1 expression is elevated in ONH in a rat glaucoma model and it is believed to mediate the astrocytic response in the glaucomatous degeneration [31, 32]. It has been reported that induction of reactive astrocytosis promotes oxidative stress in the ONH [33].
Monocytes and activated T-cells are found to accumulate in the optic nerve at the site of damage. It has been reported that monocytes migrate through the endothelium to the ONH prior to RGC degeneration in inherited mouse models of glaucoma. This infiltration may be stopped by the X-ray radiation of the eye [34] or pharmacologically and genetically [2]. Infiltration cessation acts as neuroprotection and prevents glaucoma development. These findings suggest that monocyte infiltration may be a cause of glaucoma. The activated retinal T-cells take part in the autoimmune response to the damage, probably functioning as regulatory T-cells. Plasma CD4+ T cells found in glaucoma patients present greater stimulation, proliferation and proinflammatory cytokine secretion [35].
Muller glia are the most abundant retinal glial cells. Their processes are present in all the retinal layers. They upregulate glutamine synthetase, which produces glutamate, a neurotransmitter toxic to the retina, as well as upregulating NFkB [36]. Moreover, they are known to upregulate GFAP, express TLRs, growth factors and neurotrophic factors, phagocytose debris and express various cytokines and chemokines. Muller cells have neuroprotective properties in excitotoxic damage following RGC injury [37]. It is suggested that the Muller cells may contribute to retinal damage repair by becoming pluripotent and functioning as stem cells [8].
Oxidative stress and excitotoxicity in retinal ganglion cell death
Ischemia and oxidative stress play key roles in glaucoma [31, 38, 39]. Oxidative stress induction by glial (mainly microglial) cells activation has been reported [33, 36]. Oxidative stress also affects ONH astrocytes. The cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) pathway leads to elevation in cAMP, mitochondrial dysfunction, caspase-3 activation and cell death via the AKT/Bim/Bax signaling pathway. Protein kinase A inhibition protects the ONH astrocytes from oxidative stress and increases their survival [40, 41].
Oxidative stress markers have been found not only in the retina but also in the vitreous body of glaucoma patients [42]. Natural antioxidants, such as curcumin, counteract ROS production. Curcumin was found to be neuroprotective in in vitro glaucoma models [43].
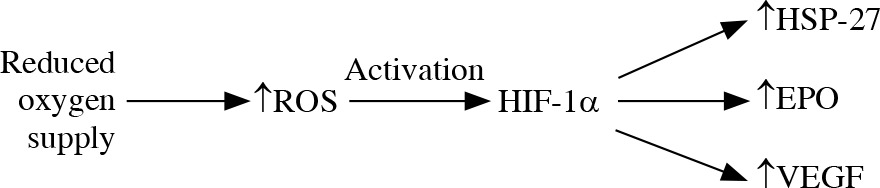
The reduced oxygen supply leads to generation of mitochondrial ROS and activation of HIF-1α that targets HSP-27, erythropoietin and vascular endothelial growth factor elevation. The location of an increased level of HIF-1α in glaucomatous eyes is connected with the location of the visual field defect location [38] (Fig. 1). It is suggested that HSP-27, although it plays a protective role in neurodegeneration by binding cytochrome c and preventing apoptosome formation, may have a direct damaging effect on RGCs. The elevation of HSP-27-reactive T cells was reported in glaucoma degeneration [44, 45]. Immunization with HSP-27 reinforces the neurodegenerative effect on S100 RGCs [14].
Fig. 1
Oxidative stress. The reduced oxygen supply leads to increase in production of reactive oxygen species (ROS) and their inefficient elimination. ROS activate HIF-1α that targets HSP-27, erythropoietin and vascular endothelial growth factor elevation
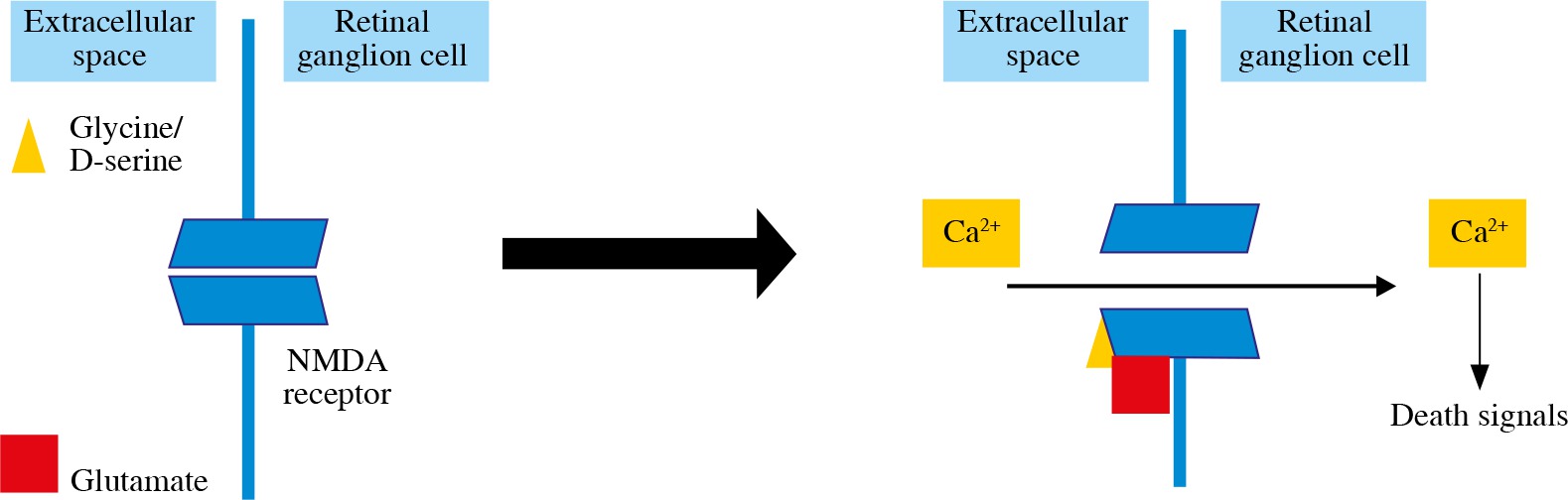
Glutamate production is the excitotoxicity factor in retinal ischemia (Fig. 2). Glutamate binds to NMDA receptors and allows the Ca2+ influx into the RGCs and apoptosis induction [31, 39]. Inducing excitotoxicity and oxidative stress with NMDA intravitreal injections is one of the RGC loss glaucomatous animal models [6]. It has been found that melanopsin-containing RGCs are not susceptible to NMDA-induced RGC loss [46]. Another glutamate receptor, AMPA-R, is also found to increase on RGCs after excitotoxic damage, glucose deprivation and oxygen/glucose deprivation, and AMPA receptor stimulation results in an increase in RGC death [36, 39].
Neurotrophic factors in glaucoma
According to the theory of target-derived deprivation of neurotrophic factors, RGC death in glaucoma is caused by a failure of retrograde axonal transport of, among other things, NTFs to the cell soma [47]. Neurotrophic factors are introduced to in vitro and in vivo glaucoma models, among others, by stem cell transplantation [48]. They play a role in pathophysiology of ophthalmic disorders other than glaucoma, such as age-related macular degeneration and Graves’ ophthalmopathy [49].
Neurotrophins, such as brain-derived neurotrophic factor (BDNF), work through cell surface receptors such as Trk and p75 receptors. TrkA binds NGF, TrkB binds BDNF and NT-4/5, TrkC binds NT-3, whereas p75 binds all the neurotrophins with similar affinity. Trk receptors play a role in cell survival, whereas p75 may play a role in cell survival as well as apoptosis [50].
Brain-derived neurotrophic factor binds to Trk receptors and activates Erk1/2 and PI3K pathways. BDNF-TrkB signaling promotes RGCs survival by upregulating neurotrophic factors that stimulate neuroprotection [51]. Erk1/2 plays a role in TrkB-induced survival of axotomized RGCs, which occurs via an MAPK-dependent pathway as a part of the intrinsic pathway of apoptosis. The finding above suggests that activation of the Erk1/2 pathway, but not the PI3K pathway, mediates TrkB-induced RGC survival [52].
TrkB expression is regulated by Shp2, which binds to growth factor receptors. Its effect has been found in RGCs exposed to oxidative stress [53]. The studies investigating the role of Shp2 in BDNF/TrkB signaling show that silencing Shp2 promotes Trk signaling and protects RGCs from susceptibility to ER stress in glaucoma [54]. The perspective of a novel gene therapy providing sustained BDNF/TrkB signaling seems promising for future application in glaucoma treatment [55].
The role of autophagy in neurodegeneration and neuroprotection
Autophagy is executed by autophagy-related genes. The RGCs’ autophagy may be inhibited by autophagy-related gene Atg4 deletion. Atg4-deficient mice showed higher p62 and lower LC3-II retina levels in the optic nerve crush glaucoma model, which suggests that the downregulation of autophagy reduces survival of RGCs [56, 57].
The Rho-associated coiled-coil-containing protein kinase (ROCK) family is the main effector of the Rho family and it comprises of ROCK1 and ROCK2. They are known for their role in RGC death. Inhibition of ROCK kinases results in increased autophagic flux seen in increased LC-II and p62 levels [58]. ROCK suppression was reported to lead to RGC regeneration, which was enhanced by the addition of CNTF [50, 58, 59]. Specific RhoA knockout was found to promote RGC survival and increase neurite outgrowth following optic nerve crush [59].
Newly introduced anti-glaucoma agents – Rho inhibitors ripasudil (Glanatec, Kowa Company, Ltd, Japan) and netarsudil (Rhopressa, Aerie Pharmaceuticals, USA) – are known inhibitors of ROCK1 and ROCK2 in a trabecular meshwork which results in decreased resistance of aqueous humor outflow and decreased IOP. Their anti-Rho activity has a neuroprotective effect on RGCs by modulating autophagy and inhibiting apoptosis [56, 60, 61]. Targeting autophagy may be a potential neuroprotective strategy in glaucoma.
Apoptosis pathways involved in retinal neurodegeneration
Intrinsic pathway
The intrinsic pathway is triggered by deprivation of the neurotrophic factor or activation of pro-apoptotic Bcl-2 family members (Fig. 3A). The TrkB receptor activates the Erk1/2 and Akt pathways that stimulate BH3-domain proteins to activate mitochondrial dysfunction [31].
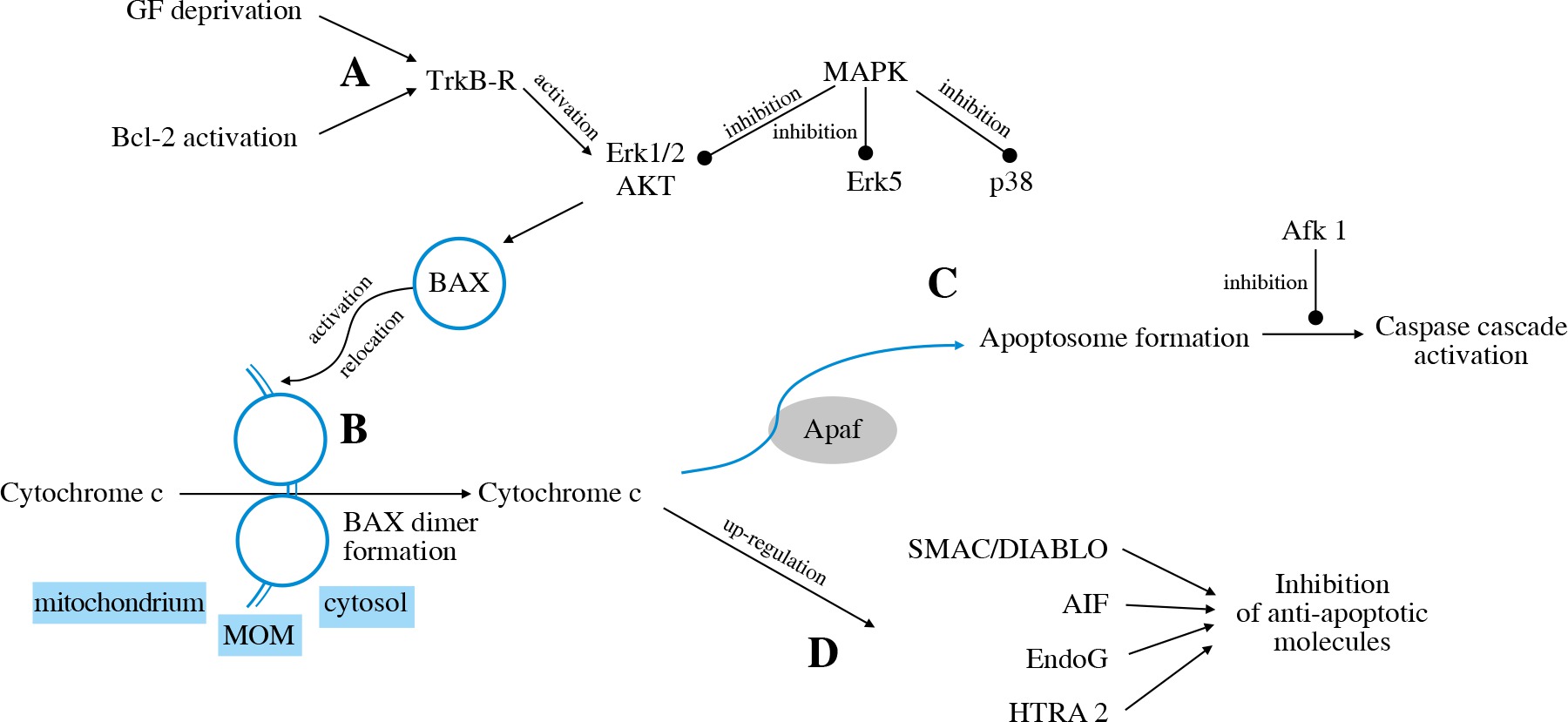
The family of BCL-2 proteins contains pro-apoptotic (e.g. BAX) as well as anti-apoptotic (e.g. Bcl-2) proteins. BAX activation, its relocation to MOM and forming dimer units lead to pore formation and release of cytochrome c into the cytosol (Fig. 3B) [62]. Cytochrome c then binds to Apaf-1, which leads to apoptosome formation (Fig. 3C) and activation of various caspases (caspase-2, caspase-3, caspase-8, caspase-9). The inhibition of caspases through Afk1 inhibition, as well as inhibition of c-Jun, attenuates RGC death [63]. It was found that targeting caspase-2 expression may also play a neuroprotective role in RGC survival [64]. Cytochrome C also activates further pathways via transmitters such as SMAC/DIABLO, AIF, EndoG, HTRA2 [32] (Fig. 3D).
The intrinsic apoptotic pathway is mediated by certain kinases: MAPKs. MKK4 and MKK7 are the MAPKs that regulate JNK. It has been found that murine RGCs deficient in Mkk4 or Mkk7 genes showed higher survival rates in glaucoma models. However, the deficiency of both Mkk4 and Mkk7 at the same time resulted in inappropriate RGC development [65].
Extrinsic pathway
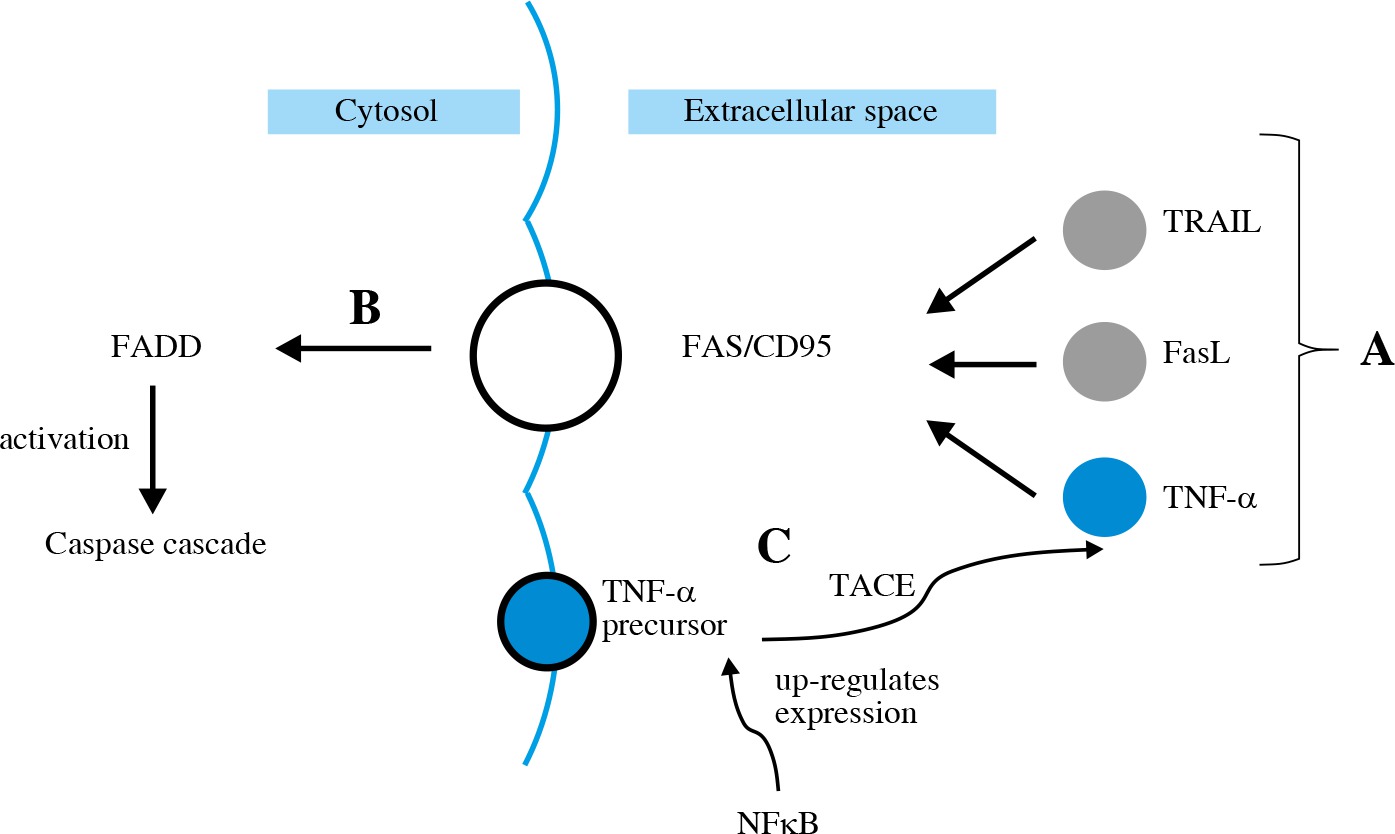
The extrinsic apoptotic pathway is induced by a specific ligand (e.g. TNF-α, FasL, TRAIL) (Fig. 4A). FasL in its soluble form, as well as TNF-α, is synthetized from a membrane-bound protein. It then binds to Fas/CD95 trans-membrane receptor which belongs to the death receptor family [31]. All these ligands activate FADD (Fig. 4B), leading to activation of caspase cascade [32]. TNF-α gene expression is up-regulated by NFkB. TNF-α production involves cleaving its precursor with the use of TACE (Fig. 4C). Then TNF-α binds to TNFR1 and TNFR2 – the death receptors.
The protective role of human antigen in glaucomatous damage
Human antigen R (HuR) protein is part of the embryonic lethal abnormal vision (ELAV) family, also called ELAV1, and is a protein that in humans is encoded by the ELAVL1 gene. This encoded protein contains 3 RNA-binding domains and binds cis-acting uracil and adenine elements.
One of its best-known functions is to stabilize mRNAs in order to regulate gene expression. It prevents neurodegeneration by controlling the oxidative metabolism in neurons [66]. It is known to promote the expression of stress response genes (Hsp70 and p53), cytokines and pro-inflammatory factors, such as TNF-α [67]. It is found in both the retina and the optic nerve. Human antigen expression is increased in the glaucomatous retina in both mice and humans. Increased HuR expression corresponds to reactive gliosis in the optic nerve [67].
Conclusions
The current glaucoma treatment works primarily by lowering the intraocular pressure. However, contemporary research underlines the importance of molecular and intracellular changes in the pathogenesis of RGC death. The knowledge of the inflammatory process taking place in glaucoma progression is vital for finding new strategies of protection against glaucoma. Gene therapy involving changes in expression of certain molecules gives hopes for developing new effective glaucoma treatment. The molecular pathways involved in neuroprotection highlighted in our review provide potential targets for innovative treatments in glaucoma.