Introduction
Cardiovascular disease (CVD) is a leading cause of morbidity and premature mortality in women and men in most of the industrialized world and many developing countries including Argentina [1]. Atherosclerosis is well established as the principal etiology of CVD, and physicians focus their effort on optimizing only the traditional risk factors (TRF) such as hypertension, diabetes mellitus, hypercholesterolemia, smoking habit, and body mass index, assuming they are the only factors that determine atherosclerosis progression.
Approximately, 50% of young patients with coronary heart disease do not have the established coronary risk factors detected in routine evaluation, and there is another category of patients in whom atherosclerosis is accelerated [2].
Serum cholesterol has been deemed a principal risk factor for atherosclerosis; this concept arose from the original Framingham Heart Study data, where the association between patients with cholesterol levels > 380 mg/dl and coronary heart disease (CHD) became clear. The same was observed when body mass index (BMI) was used in place of serum cholesterol. In 2008, it was reported that the Framingham score (FRS) based on BMI predicted cardiovascular events as effectively as FRS based on lipid profile. Many studies have confirmed this concept [3] and others have used this strategy for patient cardiovascular stratification [1, 4, 5]. This paradigm shift in cardiovascular risk relates to the potent impact of decreasing BMI in comparison to serum cholesterol levels.
The idea that lowering serum cholesterol decreases CHD was introduced by two randomized clinical trials published in The Lancet. This forever changed the course of medicine for patients with CHD. The 4S study reported a 30% mortality reduction mediated by simvastatin [6] and this reduction was associated with a 25% reduction of serum cholesterol. These beneficial effects mediated by statins were also observed with atorvastatin [7], rosuvastatin [8], and other lipid lowering drugs [9], but other reports suggest statin that benefit in CHD, does not appear to be limited to patients with marked hyperlipidemia [1].
On the contrary, the initial results from sponsored trials were not reproduced in other statin trials such as in the elderly [9, 10], in patients with heart failure [11, 12], or in patients with renal failure [13, 14]. Additionally, the Lyon Diet Heart study utilized a dietary strategy to decrease serum cholesterol and other cardiovascular risk factors. These investigators reported a 70% mortality reduction [15] without using a lipid lowering drug.
A Cochrane meta-analysis of 18 cholesterol lowering trials in patients with peripheral arterial disease also failed to demonstrate lower mortality rate [16]. The same was observed evaluating the mortality benefit in high-risk primary prevention patients [17], acute coronary syndrome patients [18, 19], or mortality outcome in the Cholesterol Treatment Trialists (CTT) evaluating low-risk patients [20].
The hypothesis that decreasing serum cholesterol would decrease atherosclerosis and cardiovascular events is very straightforward, but today, we clearly know that atherosclerosis growth is also regulated by hypertension, diabetes mellitus, BMI, smoking habits, and other non-classical risk factors [21]. This hypothesis has been recently evaluated using carotid total plaque area in patients from a vascular prevention clinic setting [22]. Spence and Solo studied the relationship of LDL cholesterol and atherosclerosis in 4,512 patients using carotid total plaque area as the burden of atherosclerosis. They found that atherosclerotic plaque did not decrease despite low levels of LDL-C (69 mg/dl), and that changes in LDL-C were not correlated with plaque progression or regression.
This evidence indicates that risk factors beyond the classical risk factors contribute to atherosclerosis growth, such as genetics, diet, glucose, L-carnitine, renal failure and toxic compounds that are renally excreted, including homocysteine, asymmetric dimethylarginine, thiocyanate, and metabolic products of the intestinal microbiome, with trimethylamine n-oxide (TMAO) and p-cresyl sulfate [23]. A recent review suggests that combining various new risk markers, such as CT-1, leptin, and resistin could help to improve risk-based intervention and to achieve the optimal therapeutic approach, but these markers are not yet extensively used in clinical practice [24].
The Framingham risk score (FRS), PROCAM, EU-SCORE, and Reynolds algorithms are commonly used risk prediction tools to stratify the risk of CVD for asymptomatic subjects. In Europe, the Systematic Coronary Risk Evaluation (SCORE) model is one of the most used [25], while in the Americas, the FRS that is based on a set of traditional risk factors including smoking, hypertension, diabetes, and lipid profile [26]. Many patients classified as low-risk by the Framingham risk score, however, still exhibit significant subclinical coronary artery disease [27], and a substantial number of patients experience CVD [27], pointing to the possibility that other characteristics not included in the FRS may help to identify high-risk patients. It has been previously shown that traditional risk algorithms underestimate cardiovascular risk in relatives of patients with premature CVD [28].
Although a family medical history of premature CVD (FHx) has been considered, the putative CVD risk factor for decades, it has not been incorporated into the most commonly used version of FRS or other widely applied multivariable risk algorithms [29–32]. This may be because the relative contribution of atherosclerosis to risk conferred by FHx has not yet been firmly established [33].
Previous studies have assessed the relationship between FHx and IMT [34] or CAC score, yet none has measured atherosclerosis burden. While IMT and CAC score are commonly used to estimate atherosclerosis, it is becoming increasingly clear that IMT does not truly represent atherosclerosis [35]. Carotid total plaque area (TPA) measured by duplex ultrasound is a highly sensitive measure of subclinical atherosclerosis and a strong predictor of cardiovascular risk [36]. Spence et al. [37] reported that in comparison to patients in the bottom quartile, patients in the top quartile of TPA had a 3.4 times higher 5-year risk of stroke, death, or myocardial infarction, after adjusting for age, sex, blood pressure, smoking (pack-years), cholesterol, diabetes, homocysteine, and treatment for blood pressure and cholesterol. They also reported that patients with plaque progression had twice the risk of those events after adjustment for the same panel of risk factors. Their findings were validated in the Tromsø study, a population-based study with over 6,000 persons; TPA, but not IMT in the distal common carotid, was a strong predictor of myocardial infarction [38] and stroke [39].
Furthermore, previous studies assessing the relationship of FHx to IMT or CAC score have included participants with traditional risk factors [40–43]. Because it is difficult to quantify risk factors such as smoking history or diabetes, multivariable adjustment for these risk factors poses a challenge, and can create poorly matched controls and unreliable results. In the current study, our objective was to eliminate the effect of these traditional risk factors and investigate the relationship between FHx and atherosclerotic burden in the absence of traditional risk factors (TRF). We then examined the same association in a hypertensive cohort with otherwise identical exclusion criteria. Finally, we used a generalized linear model to examine TPA progression with age in the setting of FHx.
Material and methods
Study participants
The study population was composed of 4,351 physician-referred individuals, participating in an atherosclerosis prevention program (LifeQualityA) conducted by Blossom DMO Argentina (2007–2014). The study was approved by the Blossom DMO Argentina ethics committee and the Western University Human Subjects Ethics Review Board (Protocol 105206). Each participant gave written consent. Exclusion criteria were based on TRF and defined as personal history of CVD, hypertension, blood pressure ≥ 130/80 mm Hg, diabetes mellitus type I or II, hypercholesterolemia, smoking history, and body mass index > 25 kg/cm2.
Following exclusion for these risk factors, 34 patients with FHx were identified. An age, sex, blood pressure, and BMI-matched control group of 56 participants was then generated by using the “hide” feature in Excel to conceal the column for TPA before systematically removing controls. The control group was sorted according to each factor, and controls were removed until the mean age, BMI, systolic and diastolic blood pressure, and percentage of males were similar to the FHx group.
To generate a second study population with hypertension as the sole TRF, we included only patients with hypertension and applied all other exclusion criteria as above. There were 32 participants with FHx identified, and an age, sex, blood pressure, and BMI-matched control group of 44 participants was created, using the same method as above.
Risk factor assessment
All individuals provided details of their demographics, medical history, and concomitant medication. A history of cigarette smoking was considered present if a subject was a current or former smoker. Patients were considered to have diabetes, hypertension, or hyperlipidemia on the basis of a self-reported diagnosis or previous use of oral hypoglycemic agents, insulin sensitizers, subcutaneous insulin, anti-hypertensive medication use, or lipid lowering drugs. Body mass index (BMI) was calculated from height and weight measurements obtained in the clinic. Blood pressure was assessed as the mean of three measurements performed on the left arm in the sitting position, after a 5-minute period of rest, with the OMRON Hem 705 sphygmomanometer [44]. Personal history of CVD was defined by prior myocardial infarction, coronary/ peripheral revascularization, stroke, or any self-reports of chest pain, chest pressure, or chest tightness. Premature cardiovascular disease was defined as a cardiovascular event occurring before the age of 51 years in men and 56 years in women, consistent with the literature on identifying a genetic predisposition [45, 46]. A positive family medical history was defined as ≥ 1 first degree and/ or ≥ 2 s degree family members with cardiovascular disease before the age of 55 years in men and 60 years in women [47]. Arrhythmias were not considered as part of the definition of premature cardiovascular disease.
Clinical risk scoring
The FRS sex-specific risk equations that substitute BMI for lipid profile (BMI-FRS) were used to predict the risk of developing CVD over the next 10 years, as previously described [44]. This traditional risk assessment score was estimated based on the subject’s reported smoking, age, sex, diabetes status, BMI, systolic blood pressure, and use of antihypertensive therapy. A previously described [4] risk calculator for CVD was used that incorporates TPA measurements into FRS to produce 10-year risks for each of stroke, myocardial infarction (MI), and all-cause CVD. The predicted risk incorporating TPA is referred as the post-test risk; it has been shown to significantly increase the AUC in ROC of risk prediction [48]. The TPA was also used alone to calculate the combined 5-year risk of stroke, myocardial infarction, and vascular death, as described previously [37].
Carotid total plaque area determination
Total carotid plaque area (TPA) was measured as described previously [44], with a high-resolution duplex ultrasound scanner. Plaque was defined as a local thickening of the intima > 1 mm in thickness. Measurements were made in magnified longitudinal views of each plaque seen in the right and left common, internal, and external carotid arteries. The sum of cross-sectional areas of all plaques seen between the clavicle and the angle of the jaw was taken as TPA.
Statistical analysis
Data were expressed as mean and standard deviation (SD) in text and tables, and as mean and standard error of the mean (SEM) in figures. Unpaired two-tailed t-tests were used to assess differences between FHx-positive and control groups. For analyses using the generalized linear model, TPA was defined as a dependent variable and FHx as an explanatory variable in order to assess whether TPA differs in the presence or absence of FHx. Given that, TPA could be described in values between 0 and ~1,200 mm2; the generalized linear model was used to assign a gamma distribution to the random component of the model using the log link function. Values of p < 0.05 were considered statistically significant.
Results
Among the 4,351 participants included in this study, 1,320 (30%) had a FHx. The average age of the participants was 58.6 ±12.5 (SD) years and 61% were male patients. The population had a relatively high prevalence of traditional risk factors: 20% of participants were diabetic, 50% were hypertensive, 56% had a positive smoking history, and 11% had personal history of CVD. Mean BMI was 25.3 ±5.5 kg/cm2, and mean blood pressure was 136.4 ±7.8 systolic and 80.7 ±11.0 mm Hg diastolic. Based on these risk factors, the mean BMI-FRS was 25.3 ±18.7% 10-year risk. The mean TPA of the participants was 58.5 ±71.5 mm2 (Table I).
Table I
Population characteristics of the Blossom DMO database (means ± SD) Argentina (2007–2014)
Cohort with no traditional risk factors
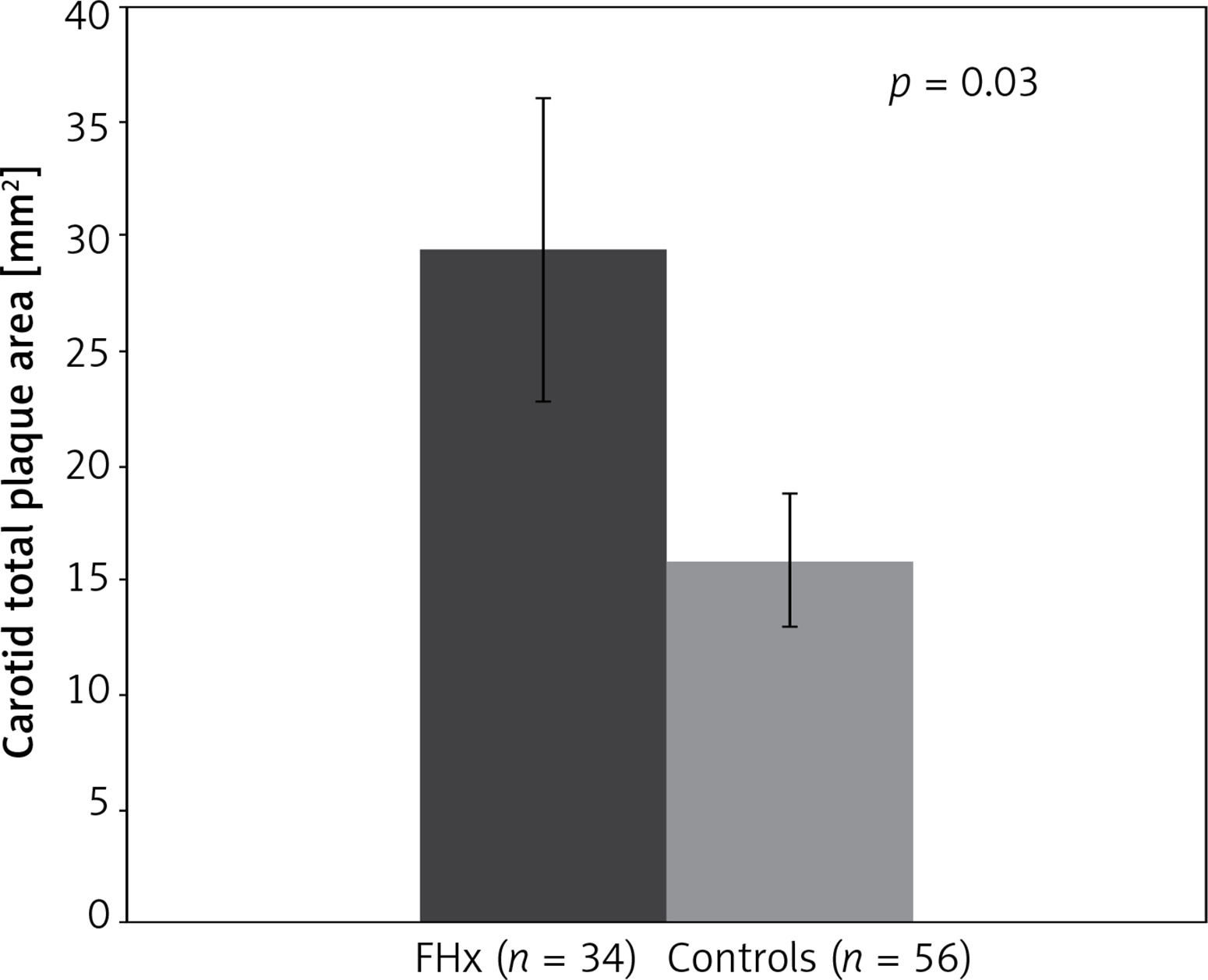
A group of 34 FHx-positive patients with no TRF were compared to an age, sex, blood pressure, and BMI-matched control group of 56 participants. FHx was associated with increased carotid plaque burden (TPA). Mean TPA was 29.4 ±38.6 mm2 for patients with FHx and 15.8 ±21.7 mm2 among controls showing that in the absence of traditional risk factors, TPA was 86% higher in patients with FHx (p < 0.05). No significant difference was observed in post-test risks for stroke, MI, or all-cause CVD (Table II, Figure 1).
Table II
Analysis of FHx-positive patients versus controls in the absence of all listed risk factors (means ± SD) Argentina (2007-2014)
| Parameter | FHx* (n = 34) | Controls (n = 56) | P-value |
|---|---|---|---|
| Total plaque area [mm2] | 29.4 ±38.6 | 15.8 ±21.7 | 0.03 |
| Framingham Risk Score (%) | 6.4 ±4.1 | 6.7 ±5.8 | 0.78 |
| Post-test risk of CVD (%) | 15.6 ±15.4 | 12.3 ±14.4 | 0.31 |
| Post-test risk of stroke (%) | 4.3 ±6.0 | 3.1 ±4.0 | 0.24 |
| Post-test risk of MI (%) | 10.9 ±11.7 | 8.6 ±10.9 | 0.34 |
| Age [years] | 53.4 ±13.5 | 53.5 ±14.8 | 0.99 |
| Male (%) | 35.3 | 35.7 | 0.97 |
| Body mass index [kg/m2] | 22.9 ±1.7 | 22.2 ±3.4 | 0.29 |
| Systolic blood pressure [mm Hg] | 116.9 ±7.7 | 116.2 ±9.2 | 0.71 |
| Diastolic blood pressure [mm Hg] | 70.7 ±5.2 | 69.0 ±5.2 | 0.13 |
| Generalized linear model* | 0.67 | 0.05 | 0.005 |
Cohort with hypertension
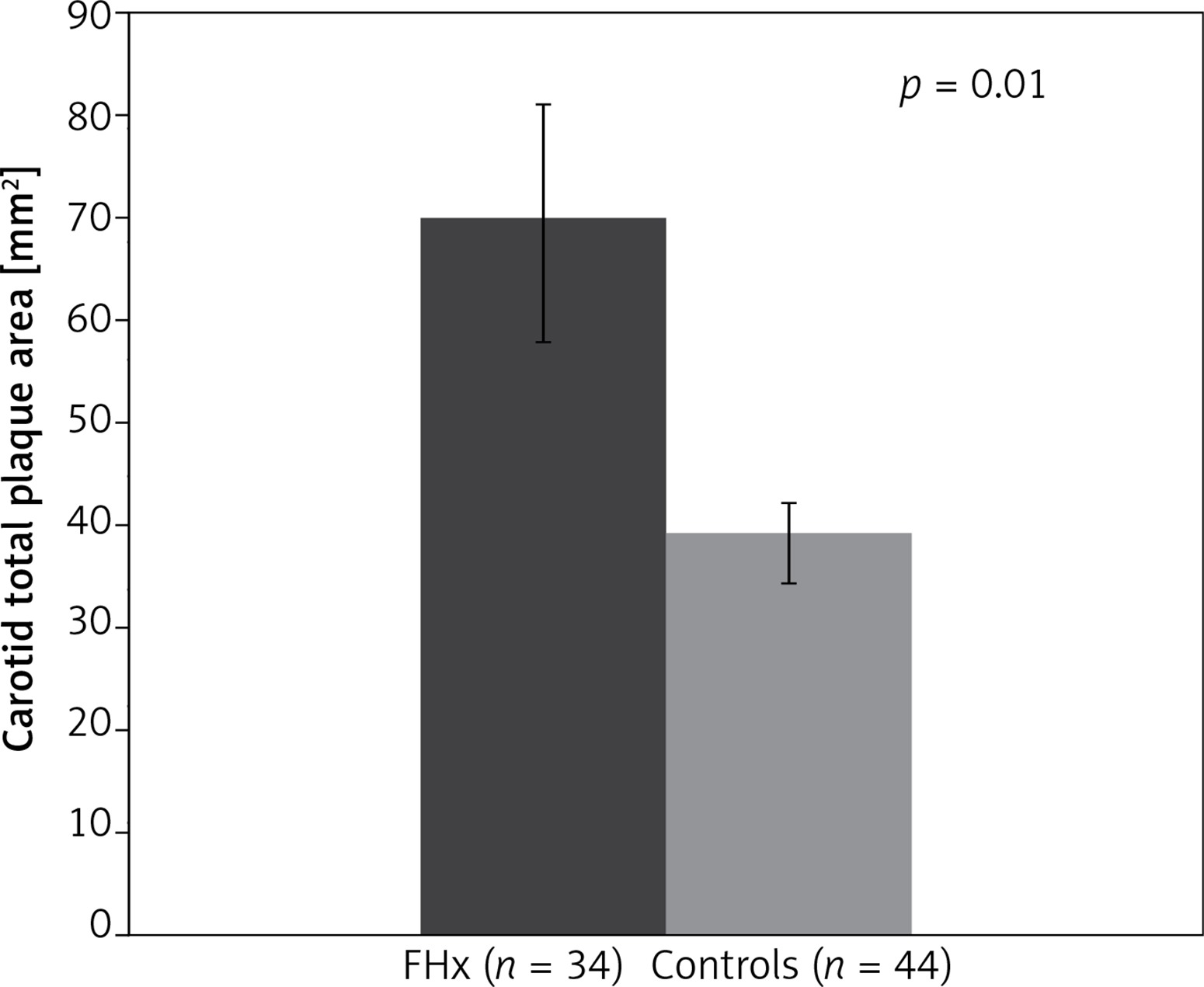
In order to further assess the association between FHx and TPA, a second study population composed of 32 FHx-positive participants with hypertension and no other TRF was compared to an age, sex, blood pressure, and BMI-matched control group of 44 participants. Mean TPA was 69.7 ±68.7 mm2 for the FHx group and 39.4 ± 35.0 mm2 for controls. Thus, in hypertensive patients with the absence of all other TRF, TPA was 77% higher in patients with FHx (p < 0.05), confirming our original analysis. No significant difference was observed in post-test risks for stroke, MI, or all-cause CVD (Table III, Figure 2).
Table III
Analysis of FHx-positive patients versus controls in the presence of hypertension and absence of all other listed risk factors (means ± SD), Argentina (2007–2014)
| Parameter | FHx (n = 32) | Controls (n = 44) | p-value |
|---|---|---|---|
| Total plaque area [mm2] | 69.7 ±68.7 | 39.4 ±35.0 | 0.01 |
| Framingham risk score (%) | 22.5 ±15.7 | 24.3 ±15.4 | 0.62 |
| Post-test risk of CVD (%) | 47.2 ±23.7 | 44.9 ±25.5 | 0.69 |
| Post-test risk of stroke (%) | 17.0 ±13.7 | 15.1 ±12.4 | 0.54 |
| Post-test risk of MI (%) | 36.5 ±21.6 | 34.4 ±21.6 | 0.67 |
| Age [years] | 66.3 ±11.6 | 66.8 ±13.1 | 0.86 |
| Male (%) | 34.4 | 34.1 | 0.98 |
| Body mass index [kg/m2] | 23.6 ±1.1 | 23.2 ±1.4 | 0.24 |
| Systolic blood pressure [mm Hg] | 139.8 ±21.3 | 144.2 ±17.1 | 0.31 |
| Diastolic blood pressure [mm Hg] | 77.8 ±12.0 | 80.3 ±10.0 | 0.33 |
| Generalized linear model* | 0.61 | 0.02 | 0.006 |
TPA progression is accelerated in the presence of FHx
Generalized linear model results suggest that TPA increased proportionally to the logarithm of age in both FHx patients and controls, and that this increase was significantly greater in the presence of FHx. For each additional year of age, TPA was found to increase by 0.67 log (TPA) (p < 0.005) in the FHx group and by 0.05 log (TPA) (p < 0.005) in the control group (Table II). Similarly, in the population of patients with hypertension as the sole CVD risk factor, TPA progression was accelerated in the presence of FHx. For each additional year of age, TPA was found to increase by 0.61 log (TPA) (p < 0.006) in the FHx group and by 0.02 log (TPA) (p < 0.006) in the control group (Table III).
Discussion
We found that in the absence of all traditional CVD risk factors, FHx-positive patients had significantly elevated TPA and TPA progression in comparison with control group patients. This difference was also observed in a cohort with hypertension as the sole traditional CVD risk factor. Atherosclerotic burden and the incidence of CVD are not negligible in patients classified as low-risk by FRS and other preferred risk scores that do not yet recognize FHx as a risk factor [49]. Our results strengthen the evidence relating FHx to increased risk of atherosclerosis, and support ultrasound screening in patients with FHx as a method to detect high-risk individuals, otherwise missed by traditional risk scores such as FRS.
A 10-year risk calculator for CVD that incorporates TPA measurements into the FRS was developed previously [4] and is referred as the post-test risk calculation. In our analyses, the mean post-test risk was not found to be significantly higher in FHx than controls, as the risk score is heavily weighted by the same FRS factors (age, sex, BMI, and blood pressure) that were balanced between the FHx and control groups. Evaluating patients individually with the post-test risk, however, revealed that the elevated TPA measured in some of our patients with no identified classical risk factor other than FHx does correspond to a clinically meaningful increase in CVD risk.
FHx-positive patient 136652 from our database illustrates this point. She is 66 years old and based on FRS has a low 10-year CVD risk of 6.03%. Accordingly, she would not require primary prevention. However, she is FHx-positive, with an elevated TPA measurement of 122 mm2. The post-test calculation revealed a migration to high-risk with a modified 10-year all-cause CVD risk of 22.1%, under these new conditions; the patient may require lifestyle changes and pharmacological treatment. This highlights a need for change in our approach to estimate a risk in FHx-positive patients. Today, routine screening for atherosclerosis in FHx-positive patients with few to no traditional risk factors is not a common practice. These results indicate that FHx-positive patients may benefit from measuring carotid TPA, because it may detect individuals misclassified as low-risk, who would benefit from primary prevention.
Although a number of studies have previously assessed the relationship between FHx and IMT [50], none has investigated this relationship in the absence of all TRF, and none has measured atherosclerosis burden. Because most cardiovascular events are not predicted by risk factor profiles [51], it is very important to have better methods to identify high-risk patients who would be likely to benefit most from preventive therapy. The current study provides new and more sensitive approach to identifying high-risk patients.
While CAC score and IMT are commonly thought to estimate preclinical atherosclerosis, it is clear that IMT is not atherosclerosis [35, 52]. The current study is the first to examine the relationship between FHx and atherosclerosis using TPA. TPA is recognized as a highly sensitive measure of subclinical atherosclerosis and a strong predictor of CVD [36, 37]. Compared with other techniques such as CAC score and IMT, TPA determination is easy, fast, and less expensive [36]. Unlike CAC score, TPA is able to identify non-calcified as well as calcified atherosclerotic plaque, making it more sensitive and permitting detection of plaque in early stages, effectively shortening the gap between detection of risk and treatment [53]. Furthermore, TPA is more strongly predictive than IMT [38, 39, 54].
The limitations of our study are that our sample was limited to patients from Argentina, where not all ethnic groups were evaluated. Additionally, misclassification of subjects by FHx may remain a potential limitation, since no verification of self-reported family medical history was performed. Lipid profile data is not a limitation based on the present bibliography demonstrating that today, many cardiovascular events occur in the presence on normal or low serum lipids. While this did limit our ability to create ideally matched controls, an updated FRS that exchanges BMI for plasma lipids has been shown to be an equal predictor of CVD risk [55].
According to previous reports [56–59], individuals with FHx tend to have an unfavorable cardiovascular risk profile, with a higher prevalence of hypertension, diabetes, or increased BMI. Furthermore, many studies investigated the associations between positive family medical history and other factors such as lifestyle [60], premature birth [61], or impaired growth in utero [62], inducing epigenetic changes and exaggerated atherogenesis in these patients that are associated with classical risk factors and situate these patients at higher risk. This may offer a partial explanation of the association between FHx and carotid plaques described in the literature. However, our results have demonstrated that FHx predisposes to atherosclerotic plaque formation even in the absence of traditional risk factors, suggesting that other possibly heritable factors are at play [63]. Further study of such heritable factors would lead to a better understanding of the familial aggregation of CVD.
Preventive treatment in FHx-positive individuals is still a matter of debate [29]. Future studies of patients and populations are warranted to assess the risks and benefits of medical interventions, such as intensive lipid-lowering therapy in persons with FHx who harbor significant subclinical atherosclerosis. Future study should also focus on how the definition of FHx can be modified to best identify individuals at risk, and whether family medical history of late-onset CVD or premature CVD in second-degree relatives significantly elevates the risk.
In conclusion, a family medical history of premature vascular disease was associated with increased carotid plaque burden in the absence of other CVD risk factors. Appropriate ultrasound screening in patients with FHx can detect high-risk patients who may benefit from early intervention.