





Abnormal values for standard laboratory tests of coagulation are frequently reported in critically ill patients. Standard laboratory tests of coagulation include activated partial thromboplastin time (aPTT), prothrombin time (PT), thrombin time (TT), fibrinogen concentration (Clauss method) and platelet count (PLT). Septic patients are frequently hospitalised in the intensive care unit (ICU) and may present with sepsis-associated coagulopathy, characterised by prolonged PT and low PLT, as well as disseminated intravascular coagulation, characterised by low PLT, elevated fibrin-related marker (soluble fibrin monomer, fibrin degradation product), prolonged PT and low fibrinogen [1]. Isolated prolongation of aPTT occurs less frequently in the ICU setting and has numerous causes. Preanalytical error should be considered in the context of isolated prolongation of aPTT in the ICU setting. It is important to mention that aPTT suffers from more interferences than PT. Factors such as reagent or activator used may have an impact here; therefore clinical laboratories should establish local reference values for aPTT [2]. Apart from preanalytical error, acquired and congenital causes may lead to isolated prolongation of aPTT. Herein, we discuss the rationale for aPTT testing, limitations of aPTT, a practical algorithm for differential diagnosis of isolated prolongation of aPTT in the ICU, and management of disorders associated with isolated prolongation of aPTT based on the current literature.
ACTIVATED PARTIAL THROMBOPLASTIN TIME
Activated partial thromboplastin time has been historically ordered as a screening test for coagulation factor deficiencies [3]. It measures the time in seconds that is required for formation of a fibrin clot in a platelet-poor plasma sample, to which appropriate amounts of calcium chloride (a process called recalcification), reagent (historically cephalin – extract of rabbit brain tissue rich in phospholipids) and activator (negatively charged particulate substances, e.g. kaolin, silica, ellagic acid, celite) are added. It is sensitive to significantly lowered activities of intrinsic system (factors XI, IX, VIII), common system (factors X, V, II, fibrinogen) and contact system (factor XII, kallikrein, high molecular weight kininogen – HMWK) components. Reagent used for aPTT usually lead to prolongation when activity of factors VIII, IX, XI is reduced to ≤ 30% and fibrinogen concentration is < 100 mg dL-1 [4]. Low fibrinogen concentration leads to prolongation of aPTT, but also prothrombin time and thrombin time. The coagulation cascade and standard laboratory tests of coagulation are presented in Figure 1. Isolated prolongation of aPTT is caused by deficiencies of intrinsic and/or contact system components and only these will be discussed in this review article. Activated partial thromboplastin time has been used since1970 to monitor anticoagulation with unfractionated heparin (UFH) [5] and some direct thrombin inhibitors, but there are several factors that may impact on an aPTT result: reagent used, activator used, UFH composition, UFH concentration, coagulation factor deficiencies, and coagulation inhibitors [6, 7]. Use of anti-factor Xa assay may overcome these limitations [8, 9], but it may not be so in an acute phase reaction [10]. Lupus anticoagulant (LA), as an antiphospholipid antibody, partially neutralizes reagent phospholipids and may also lead to isolated prolongation of aPTT. Reagents used for the aPTT test differ with respect to responsiveness to LA (high, intermediate, low) and clinical laboratories should select aPTT reagents based on their most common clinical indications (anticoagulation monitoring, coagulopathy screening, LA detection) [4].
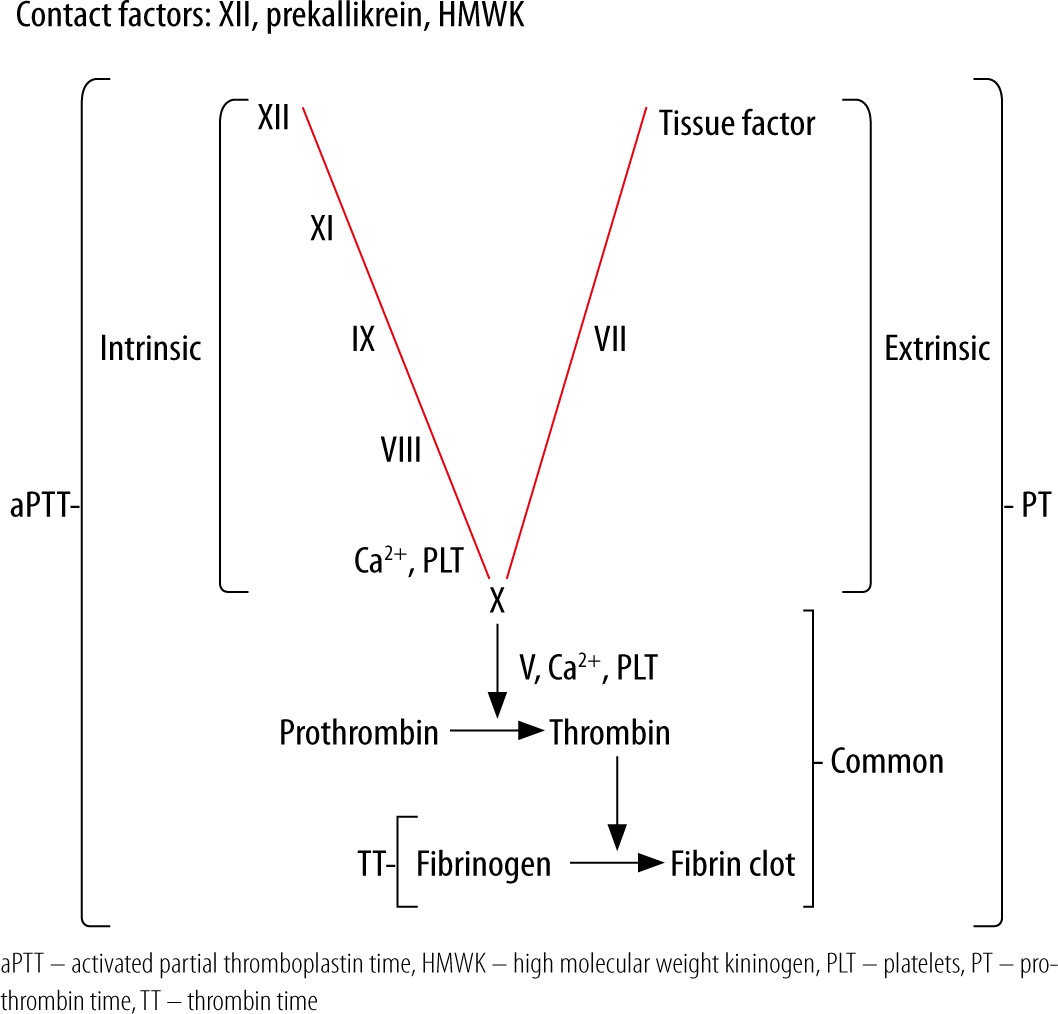
Prolonged aPTT in the absence of clinically relevant bleeding is a common finding in patients hospitalised in the ICU [11]. When aPTT is prolonged, it is crucial to distinguish between normal haemostasis and bleeding tendency, as it may lead to decreased doses of anticoagulants, withdrawal of anticoagulants, or even administration of pro-haemostatic medications. This is especially valid in critically ill subjects, in whom the prevalence of thrombosis is high, reaching 32% in a recent study [12]. On the other hand, shortened aPTT is mostly due to preanalytical error, although in small percentage of patients it may predict thromboembolic complications (mainly due to high concentrations of factor VIII and fibrinogen) [13]. To sum up, prolonged aPTT does not predict bleeding or thromboembolic complications; hence interpretation of an aPTT result constitutes a daily challenge to the ICU personnel.
DIFFERENTIAL DIAGNOSIS OF ISOLATED PROLONGATION OF activated partial thromboplastin time
1. Preanalytical errors
(a) Haemolysis, hyperbilirubinaemia, hypertriglyceridaemia
The complexity of haemostasis and analytical methods employed in coagulation testing makes them more susceptible to interferences from abovementioned contaminants [14]. Haemolysis is an important interfering factor here. The aPTT is measured using electromechanical or optical techniques [15]. Haemolysis interferes with both methods either through cell-free haemoglobin absorbance at wavelengths used in optical methods (optical interference) or though release of molecules activating coagulation and platelets (biologic interference) [14]. Hypertriglyceridaemia might also activate coagulation. Both hypertriglyceridaemia and hyperbilirubinaemia interfere with optical methods. Clinicians should reflect on these possible interferences when facing patients with elevated cell-free haemoglobin, triglycerides, and bilirubin. Discussion with clinical laboratory personnel is advised.
(b) C-reactive protein
C-reactive protein (CRP) interferes with aPTT assays and causes prolongation of aPTT in a concentration- and essay-dependent manner. This interference is most likely phospholipid-dependent. When an isolated prolongation of aPTT is found in patients with inflammation, it is advocated to repeat the test with a CRP-independent assay [16, 17].
(c) Blood clot in a test tube
The presence of a clot in a test tube leads to prolongation of aPTT due to consumption or activation of fibrinogen [18].
(d) Ratio of blood to anticoagulant
An adequate amount of blood should be placed in a test tube for standard laboratory tests of coagulation. The optimal ratio of blood to anticoagulant is 9:1. Haemoconcentration was demonstrated to prolong aPTT in one study, emphasizing the importance of the blood-to-anticoagulant ratio [19].
2. Acquired causes
(a) Lupus anticoagulant without antiphospholipid syndrome
Presence of lupus anticoagulant (LA) was found to be the most common cause of isolated prolongation of aPTT in different populations of critically ill patients: acute care (53.1%) [20], and post kidney transplantation (68.4%) [21]. LA is associated with sepsis and/or catecholamine treatment, hence is frequently present in critically ill subjects. LA was also the most frequent cause of prolongation of aPTT in an outpatient haemostasis clinic – in 22.6% of cases [22]. The prolonged aPTT in this setting is not associated with bleeding or thromboembolic complications [23]. It is important to distinguish between isolated presence of LA and presence of LA as a laboratory manifestation of antiphospholipid syndrome (APS) in which thrombosis or spontaneous abortion may occur [24].
(b) Antiphospholipid syndrome
The classical APS is characterised by the presence of antiphospholipid antibodies which bind target phospholipid molecules, mainly through β2-glycoprotein I (β2GPI), and are associated with recurrent foetal loss and thromboembolic complications [25]. A subset of the APS with high mortality termed catastrophic APS was first described in 1992 [26]. Triggering factors for a catastrophic APS include factors frequently present in ICU patients: infection, trauma, anticoagulation withdrawal, and various carcinomas [27]. The prolonged aPTT in APS is associated with thromboembolic complications and requires therapeutic anticoagulation.
(c) Contact system factor deficiency
Frequency of acquired factor XII deficiency varies among populations. In one ICU study, as many as 50.6% of patients with isolated aPTT prolongation had factor XII deficiency (80% of these patients did not experience a higher bleeding rate) [11], whereas in another study in a general acute care hospital population, deficiency of factor XII was a rare cause of prolonged aPTT [20]. Deficiency of factor XII was found in 75% of patients with isolated prolongation of aPTT scheduled for elective cancer surgery [28]. In factor XII deficiency prolongation of aPTT is significant, from our experience often reaching 100-200 s. Factor XII deficiency is a surrogate of contact pathway activation in this setting. There are no symptoms of a bleeding disorder. Patients with prekallikrein deficiency also remain asymptomatic [29].
(d) Acquired von Willebrand syndrome
An acquired form of von Willebrand syndrome (AvWS) results from other medical conditions: auto-immune disorders, monoclonal gammopathies, paraproteinaemias, malignancies (lymphoproli-ferative diseases, Wilms tumour, haemangiomas), cardiovascular conditions with aberrant turbulence (aortic stenosis, pulmonary stenosis, mitral valve prolapse, ventricular septal defect, prosthetic heart valve, left ventricular assist device). The various types of AvWS present with varying degrees of bleeding tendency: easy bruising, epistaxis, bleeding gums, heavy menstrual bleeding, peripartum bleeding, haemarthrosis and internal bleeding (AvWS type 3). The confirmatory test is von Willebrand factor (VWF) activity and ristocetin cofactor activity. Treatment options include underlying disease treatment, immunosuppression (corticosteroids, cyclophosphamide), intravenous immunoglobulins, and therapeutic plasma exchange. Haematology consultation is strongly advised.
(e) Acquired haemophilia A
It is a rare autoimmune disorder, with annual incidence of approximately 1 case per million population, in which there is abrupt production of autoantibodies directed against coagulation factor VIII. There is no previous personal or family history of bleeding disorders. In half of cases another condition or medication is present: lupus, rheumatoid arthritis, multiple sclerosis, Sjögren’s syndrome, temporal arteritis, inflammatory bowel disease, infection, diabetes, respiratory disease, dermatological disease, haematological cancer, pregnancy (mainly post-partum), penicillin, interferon. The remaining cases are idiopathic. Main symptoms include muco-cutaneous bleeding: ecchymoses, gastrointestinal bleeding, melaena, haematuria, and muscle haematoma [30]. In the context of severe bleeding invasive and surgical procedures are contraindicated, medications with anticoagulant and antiplatelet actions should be withdrawn, and measures to stop bleeding should be implemented. Prior to administration of blood products and coagulation factors, it is necessary to obtain a blood sample for a mixing study. The management strategy in acquired haemophilia A (AHA) is twofold: prophylaxis/control of bleeding and inhibitor eradication. There is no correlation between factor VIII activity and degree of bleeding as in congenital haemophilia A; therefore the optimal way to control bleeding is the use of factor VIII bypassing agents: recombinant activated factor VII (rFVIIa), activated prothrombin complex concentrate (aPCC), recombinant porcine factor VIII, but not human factor VIII. The clinical efficacy of the first two agents has been confirmed in clinical trials [31, 32]. Unfortunately, there are no laboratory tests to monitor therapy with rFVIIa or aPCC. If there is no clinical improvement with one of these drugs, alternating sequential therapy with the two medications may be attempted. Drugs with uncertain efficacy include desmopressin and human factor VIII concentrate. Tranexamic acid (TXA) is a supportive treatment option. Along with haemostatic agents, inhibitor eradication therapy with immunosuppressive drugs should be started. In mild/moderate bleeding, inhibitor eradication is attempted first [33]. Haematology consultation is strongly advised.
3. Congenital causes
(a) Von Willebrand disease
Von Willebrand disease is the most common congenital bleeding disorder, affecting up to 1% of the general population. Von Willebrand factor (vWF) binds to factor VIII (prolongation of half-life, secondary haemostasis), as well as collagen in the subendothelial matrix and PLT (primary haemostasis). Patients who are symptomatic have mucosal bleeding and postsurgical bleeding. The most common presentation in men is epistaxis, whereas in women it is menorrhagia. Severe cases might bleed from the gastrointestinal tract and into muscles and joints. Some individuals with mild disease may be asymptomatic and have normal aPTT. As aPTT is often normal, the diagnostic workup includes vWF activity and ristocetin cofactor activity. Most minor bleeding events do not require treatment. Mucosal bleeding may require antifibrinolytic therapy (e.g. TXA) and/or desmopressin. Invasive procedures may require vWF concentrate or PLT. Haematology consultation is strongly advised.
(b) Haemophilia A, haemophilia A with inhibitors
Usually there is a family and personal history of superficial (from abrasions, lacerations) and deep tissue (joints, muscles, gastrointestinal system, brain) bleeding episodes. Factor VIII deficiency was found in 25% of patients with isolated aPTT prolongation scheduled for cancer surgery [28]. Factor VIII deficiency is associated with bleeding complications. Sixty percent of patients have severe disease (factor VIII activity < 1%). The mainstay of therapy is with plasma-derived or recombinant factor VIII concentrates. Individuals with mild disease may have normal aPTT and be managed with desmopressin only. Haematology consultation is strongly advised.
(c) Haemophilia B, haemophilia B with inhibitors
There is factor IX deficiency. Family and personal history reveal easy bruising, haematuria, epistaxis, and haemarthrosis. Severity of bleeding depends on residual factor IX activity. Sixty percent of patients have severe disease (factor IX activity < 1%). The mainstay of therapy is with plasma-derived or recombinant factor IX concentrates. Individuals with mild disease may have normal aPTT and be managed with an antifibrinolytic agent only (e.g. TXA). Haematology consultation is strongly advised.
(d) Factor XI deficiency
Factor XI deficiency was found in 20.5% of patients with isolated aPTT prolongation scheduled for cancer surgery [28]. There are symptoms of a bleeding disorder.
(e) Factor XII deficiency
Congenital factor XII deficiency (Hageman factor deficiency) is a rare disorder that has been sporadically reported worldwide [34, 35].
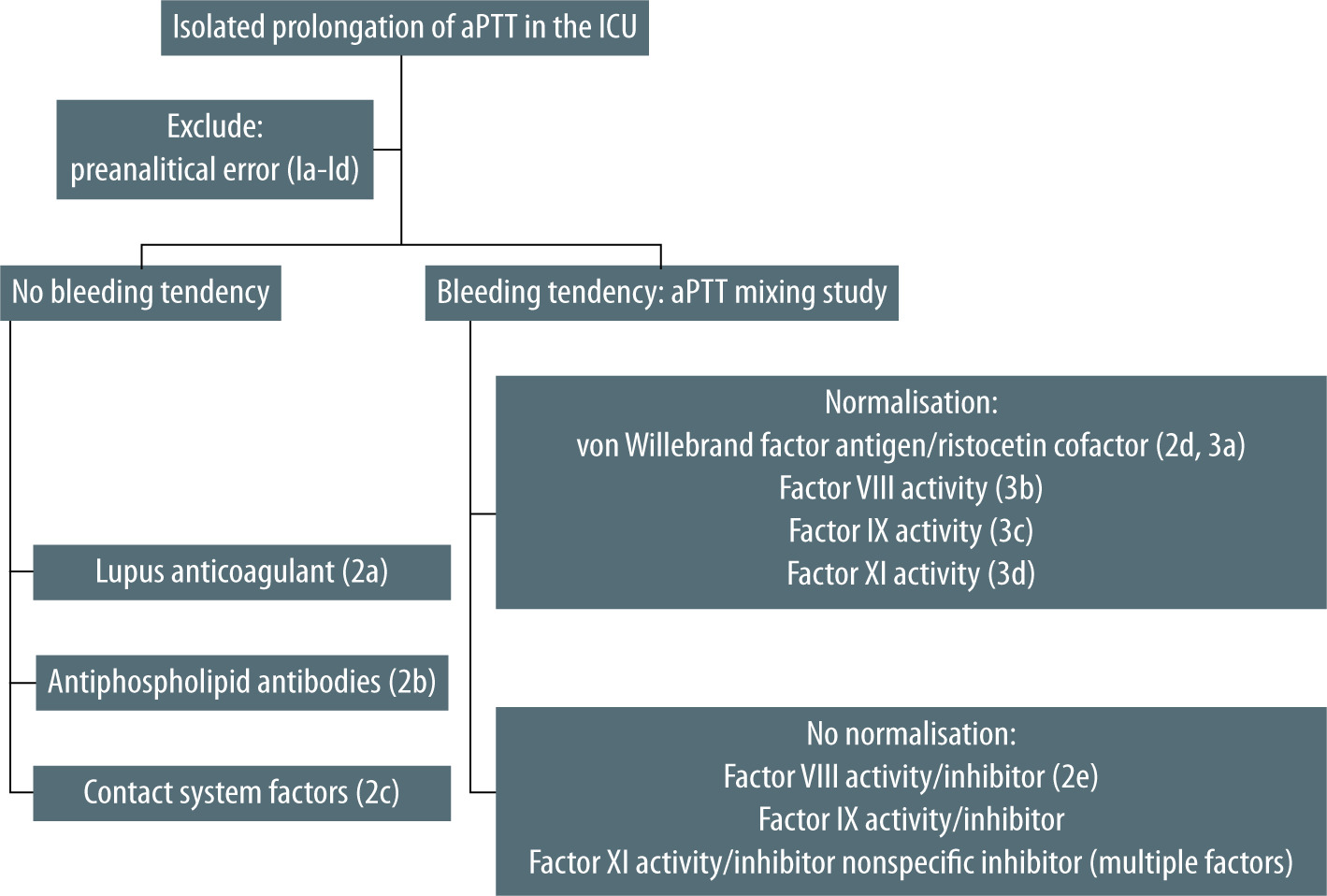
In the case of a bleeding tendency, it is clinically sound to first conduct an aPTT mixing study in which patient plasma is mixed with the normal plasma pool in a 1 : 1 ratio, following which aPTT of the mixture is determined immediately and after one hour of incubation at 37°C. Immediate and post-incubation normalisation of aPTT in the mixing study suggests deficiency of one of the intrinsic system coagulation factors (factors VIII, IX, XI). In the case of a non-bleeding tendency and similar correction pattern, deficiency of factor XII is likely. Immediate correction of aPTT and prolongation after incubation is suggestive of factor VIII inhibitor – factor VIII activity should be measured, and if low factor VIII inhibitor is determined or in non-bleeding tendency of weak LA, LA tests should be ordered. Incomplete normalisation of aPTT immediately following mixing and post-incubation is suggestive of inhibitors to factor VIII, IX, XI – activity of factors should be measured and if low appropriate factor inhibitor should be determined; in non-bleeding tendency it is suggestive of inhibitor to factor XII and LA – factor XII activity and LA tests should be ordered. The definition of correction varies between laboratories and attempts at standardisation have been unsuccessful. Great care should be taken to not contaminate the specimen with heparin. If such contamination cannot be excluded, the sample should be treated first with heparinase and the aPTT test repeated [36]. The test can be performed in a local laboratory, as opposed to confirmatory tests, which are usually send-out tests.
Based on abovementioned causes of isolated prolongation of aPTT and its clinical manifestations in ICU patients, we proposed a practical diagnostic algorithm (Figure 2).
CONCLUSIONS
As a first step in differential diagnosis of isolated prolongation of aPTT in intensive care unit patients, preanalytical error should be excluded. Next, in the case of normal haemostasis, lupus anticoagulant, antiphospholipid antibodies and contact system components should be tested for. In the case of bleeding, a locally performed aPTT mixing study allows for differentiation between coagulation factor deficiency and presence of an inhibitor. The proposed diagnostic algorithm may expedite the diagnosis with adequate management following.