Introduction
Endothelial cells (ECs) are reported to be capable of switching to a fibro-proliferative mesenchymal phenotype through endothelial-to-mesenchymal transition (EndMT) [1]. This transition process could be characterized by loss of cell-to-cell adhesion and changes in cell polarity, which in turn aggravate cell proliferation, migration and secretion of extracellular matrix proteins [2]. The expression of EC markers, such as cadherin 5 (VE-cadherin) and platelet/endothelial cell adhesion molecule 1 (PECAM1), is decreased, while the expression of mesenchymal cell markers, such as α-smooth muscle actin (α-SMA), myosin heavy chain 11 (Myh11) and calponin, is elevated [2, 3]. Shear stress and TGF-β have been regarded as the pivotal stimuli in driving EndMT progression [4, 5]. Functionally, EndMT progression is initially observed to occur during embryogenesis, where it plays an important role in cardiac valve formation [6]. Recent studies also conclude that EndMT is involved in a number of pathological conditions such as myocardial infarction, neointimal formation, pulmonary hypertension and atherosclerosis [2, 5, 7–10], whereas the exact underlying mechanism of EndMT during the pathogenesis remains poorly defined.
MicroRNAs (miRNAs) has been defined as one of the well-characterized non-coding RNAs ranging in size from 19 to 25 nucleotides. Over the past decades, a number of studies have demonstrated the important roles of miRNAs in endothelial dysfunction and EndMT progression [7]. On the other hand, long non-coding RNAs (lncRNAs) have been referred to as the transcripts longer than 200 nucleotides but without protein-coding potential. Accumulating evidence has indicated that lncRNAs are involved in a variety of biological functions such as epigenetic and post-transcriptional regulation in cellular homeostasis, cell proliferation and differentiation [11, 12]. Furthermore, lncRNAs exert sponge-like effects on numerous miRNAs, eventually regulating miRNA expression and their downstream mRNA functions. Likewise, miRNAs and lncRNAs have recently been recognized as candidate biomarkers for diagnosis, prognosis and therapeutics in cancer and cardiovascular diseases [11]. Collectively, the research relevant to lncRNA-miRNA-mRNA axes in the pathogenesis of cardiovascular diseases has been constantly reported and gradually evolving [13].
Current epidemiologic studies have consistently revealed that smooth muscle and endothelial cell-enriched migration/differentiation-associated lncRNA (SENCR) is particularly abundant in vascular smooth muscle cells (VSMCs) and ECs [14]. Despite the lack of data supporting the association between genetic polymorphisms within SENCR and the incidence of coronary artery disease (CAD), further research suggested that SENCR was involved in the regulation of VSMC proliferation and migration through controlling contractile gene myocardin and α-SMA expression, ensuing contributing to the pathogenesis of CAD [15, 16]. In the present study, we sought to investigate whether lncRNA SENCR contributes to EndMT progression and to decipher the underlying mechanism.
Materials and methods
Human samples
Carotid plaques were obtained from 5 patients undergoing endarterectomy. Healthy carotids were obtained from 3 patients undergoing trauma surgery without carotid plaques. All samples were immediately stored in liquid nitrogen after surgical removal.
Bioinformatics
The nucleotide sequence of miR-126a was obtained from the miRBase database (http://www.mirbase.org/). The candidate miR-126a binding sites within the 3′UTR was predicted by Target-Scan (http://www.targetscan.org).
Cell culture
Human coronary endothelial cells (HACECs) were purchased from Lonza Walkersville (Cat# CC02585, USA) and regularly cultured in EBM-2 medium (Clonetics, Lonza, Switzerland) supplemented with human recombinant epidermal growth factor (hEGF, 3 ng/ml, 0.1% in EBM-2), hydrocortisone, human recombinant fibroblast growth factor-beta (hFGF-b, 1 ng/ml, 0.1% in EBM-2), vascular endothelial growth factor (VEGF, 0.1% in EBM-2), insulin-like growth factor-1 (IGF-1, 0.1% in EBM-2), ascorbic acid, 5% fetal bovine serum (FBS), 1% penicillin and streptomycin and 5% CO2 at 37°C. The cells were tested positively for CD31, vWF and acetylated low-density lipoprotein uptake according to the manufacturer’s instructions (Clonetics, Lonza, Switzerland). To validate the repeatability and reliability, we purchased and applied two batches of HACECs in the following experiments. EndMT was initiated by maintaining HACECs in complete EBM-2 medium, followed by addition of 5 ng/ml TGF-β1 (Peprotech, USA).
RNA interfering and miRNA mimics transfection
HACECs were plated in six-well plates and maintained overnight. HACECs were transfected with control siRNA or SENCR siRNA and miR-126a mimics (Genepharma, Shanghai, China) at a final concentration of 50 nM using Lipofectamine 2000 (Invitrogen, USA) for 48 h. The following experiments were conducted after 48-hour transfection.
Quantitative RT-PCR
Quantitative real-time reverse transcription (RT-PCR) was applied to determine the RNA expression levels of SENCR, miR-126a, VE-cadherin, PECAM1, α-SMA, calponin, Smad ubiquitin regulatory factor 2 (SMURF2) and GAPDH. The primers predesigned for RT-PCR are listed in Table I. The amplification and detection of specific products were performed in triplicate using the SYBR Premix Ex Taq assay (Takara, Japan), and the relative expression levels between groups were calculated using the following equation: relative gene expression = 2-(ΔCtsample – ΔCtcontrol).
Table I
Summary of primer sequences used for RT-PCR
Western blot analysis
HACECs were re-suspended in lysis buffer and 2 mg/ml protease inhibitor cocktail (Roche Diagnostics Corp). Protein concentrations were determined using the Bradford Protein assay kit (Beyotime Biotechnology, China). An equal amount of protein was analyzed by SDS-PAGE and immunoblotting. Primary antibodies used included the following: α-SMA (ab5694, abcam, USA), calponin (ab46794, abcam, USA), VE-cadherin (Cat. 2500, Cell Signaling Technology, USA), PECAM1 (sc-71872, Santa Cruz, USA), SMURF2 (GTX31571, GeneTex, USA) and GAPDH (ab8245, abcam, USA). Secondary antibodies were fluorescence-labeled antibodies. Bands were visualized using the Odyssey Infrared Imaging System (LI-COR Biotechnology).
Dual luciferase assay
HEK293T cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin and streptomycin. The oligonucleotide fragments of lncRNA SENCR that contained the putative miR-126a binding sites were amplified and inserted into the Dual-Luciferase Reporter plasmid psiCHECK-2 (Promega, USA). For the luciferase assay, HEK293T cells were transfected at 80% confluency in 24-well dishes with psiCHECK-SENCR and miR-126a mimics using Lipofectamine 2000 (Invitrogen, USA) for 24 h. Likewise, SMURF2 3′UTR was cloned into the dual-luciferase reporter plasmid psiCHECK-2. Constructs carrying the mutated fragment of the SMURF2 3′UTR without the putative miR-126a binding sequence served as the mutated control. HEK293T cells were co-transfected with psiCHECK-SMURF2-3′UTR (Luci-SMURF2-WT) or with psiCHECK-SMURF2-3′UTR mutant (Luci-SMURF2-Mut), and miR-126a mimics using Lipofectamine 2000 for 24 h. Based on the Dual-Luciferase Reporter Assay System (Promega, USA), the relative luciferase expression normalized to Renilla activity was measured using an automatic microplate reader.
Statistical analysis
Statistical analysis was performed using IBM SPSS software (Version 14.0, USA). Data were presented as mean ± standard deviation (SD). The analysis of raw data was performed using Student’s t test for two groups. One-way analysis of variance (ANOVA) was used to compare multiple groups, if appropriate, with Bonferroni correction for the post-hoc analysis. Differences were regarded as statistically significance for p < 0.05.
Results
SENCR was decreased in carotid plaques and in response to TGF-β1 stimulation
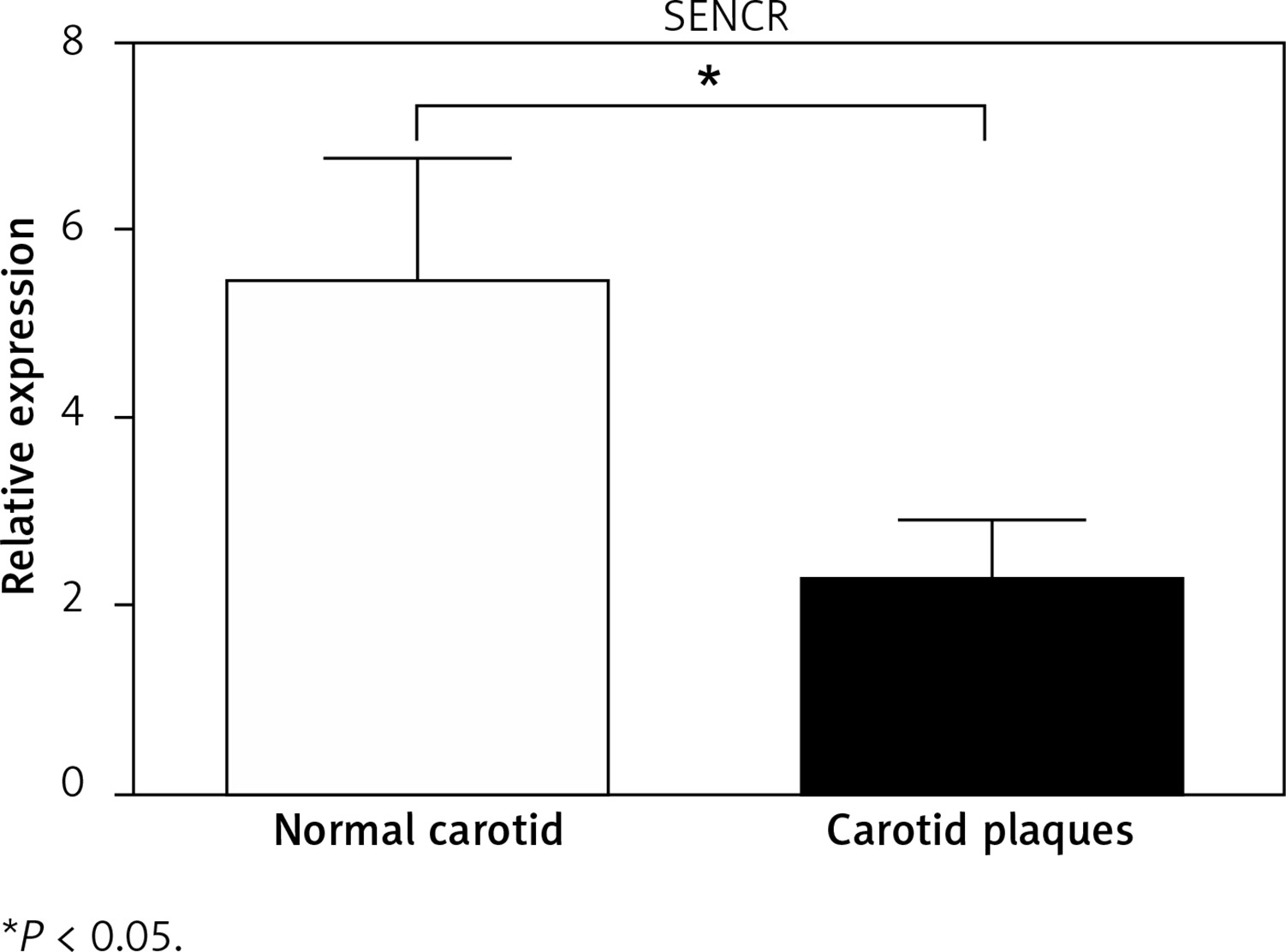
To determine the potential role of SENCR in atherosclerosis, quantitative RT-PCR was first performed to compare the expression levels of SENCR in normal carotid artery and carotid plaques. As illustrated in Figure 1, SENCR was significantly down-regulated in carotid plaques as compared to normal carotids.
Knockdown of SENCR enhanced EndMT progression of ECs
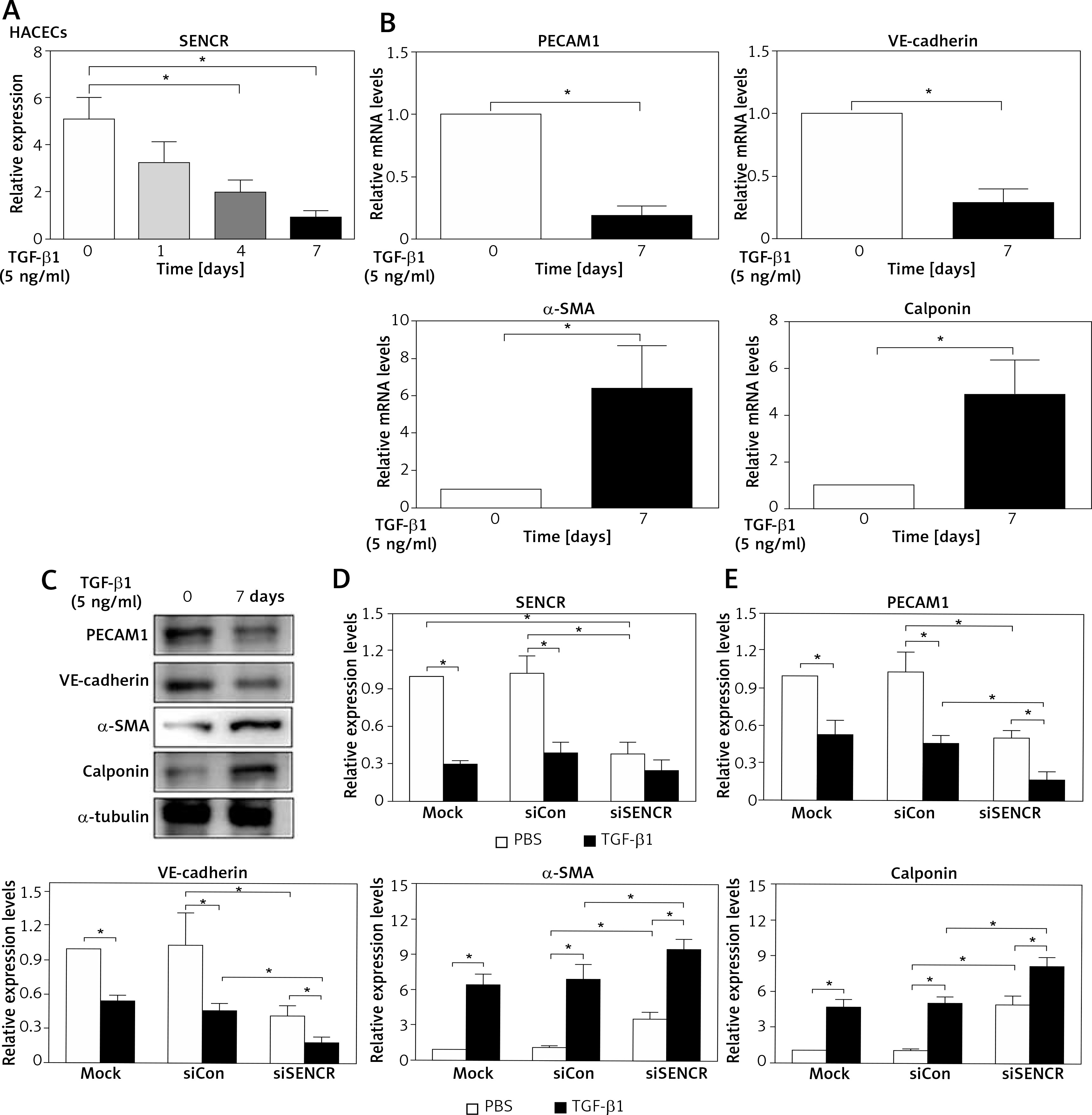
Indeed, it is widely accepted that researchers applied HACECs in vitro to validate the observations in coronary and carotid arteries, especially plaques and aneurysm. Consistent with the observation in carotid samples, SENCR expression was significantly reduced in HACECs after the stimulation of TGF-β1, which is considered as a potent atherogenic stimulus (Figure 2 A). After induction of HACECs with TGF-β1, HACECs displayed remarkably decreased mRNA and protein levels of the endothelial markers PECAM1 and VE-cadherin, accompanied by significantly increased levels of smooth muscle markers α-SMA and calponin, confirming that HACECs underwent EndMT (Figures 2 B, C). Based on the in vitro EndMT model, we then examined the effect of SENCR silencing on EndMT progression. The results showed that depletion of SENCR markedly aggravated the expression of smooth muscle markers α-SMA and calponin induced by TGF-β1 but repressed the expression of endothelial markers PECAM1 and VE-cadherin down-regulated by TGF-β1, suggesting that knockdown of SENCR aggravated EndMT progression (Figures 2 D, E).
Figure 2
Knockdown of SENCR enhances EndMT progression and inhibits tube formation in ECs. A – Quantitative RT-PCR showed that the expression of SENCR is decreased after TGF-β1 stimulation (5 ng/ml). B – mRNA expression levels of PECAM1, VE-cadherin, α-SMA and calponin in HACECs in response to TGF-β1 (5 ng/ml) for 7 days are determined by quantitative RT-PCR. C – Western blotting of PECAM1, VE-cadherin, α-SMA, calponin and α-tubulin in HACECs with TGF-β1 (5 ng/ml) stimulation for 7 days. D – Expression levels of SENCR in HACECs transfected with control siRNA (siCon) or SENCR siRNA (siSENCR) in the presence of TGF-β1 (5 ng/ml) for 4 days. E – mRNA expression levels of PECAM1, VE-cadherin, α-SMA and calponin in HACECs transfected with control siCon or siSENCR in the presence of TGF-β1 (5 ng/ml) for 4 days. Experiments were performed three times. Bars indicate mean ± SD
SENCR inhibited and bound to miR-126a
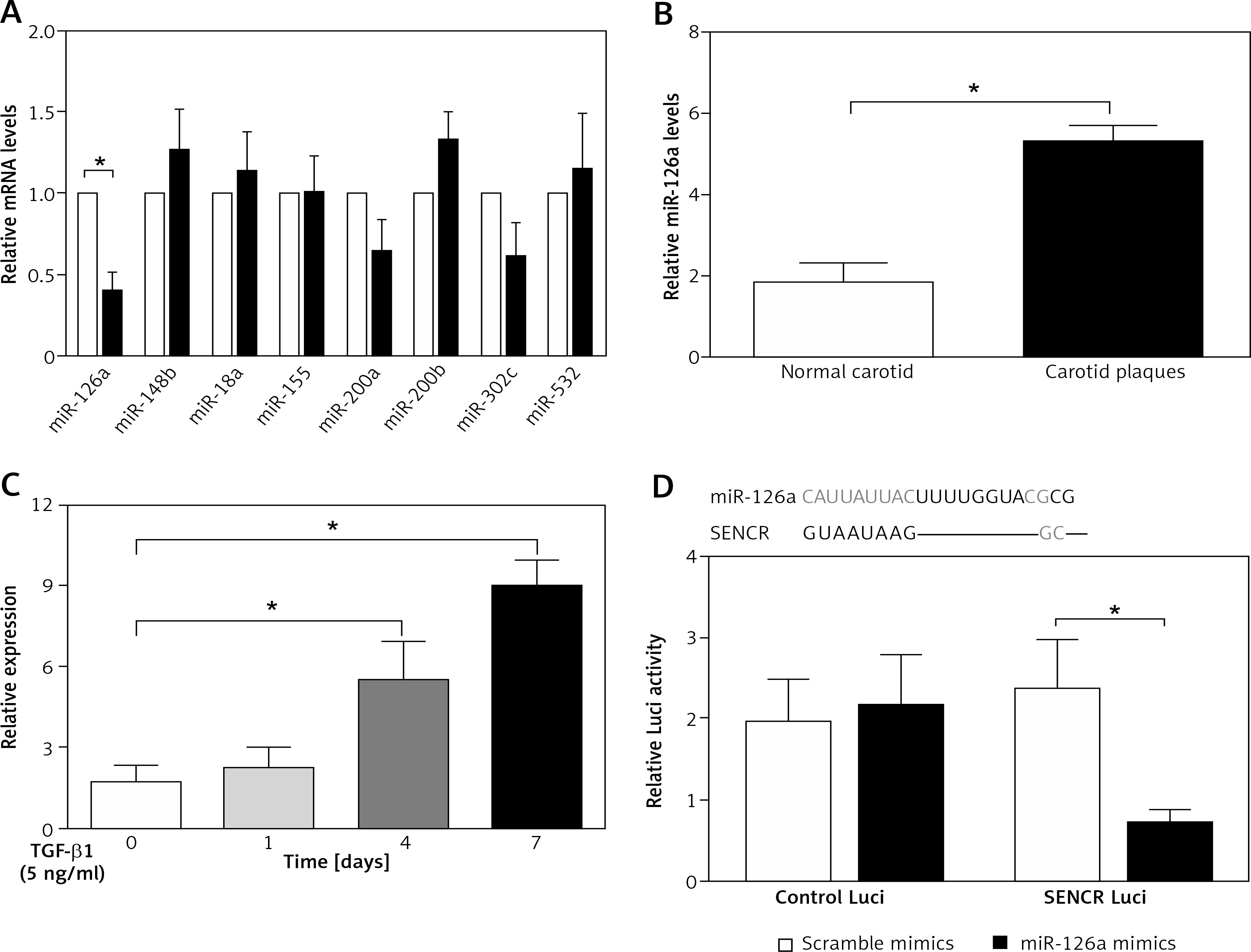
To find out the mechanism underlying the regulation of SENCR on EndMT and investigate the interplay between SENCR and miRNAs, we transfected HACECs with SENCR siRNA (siSENCR) and determined the miRNA targets based on previous reports. Among the candidate miRNAs, miR-126a emerged as a particularly interesting candidate that was decreased by 63.7% in HACECs transfected with siSENCR as compared to the control group (Figure 3 A). Likewise, the expression of miR-126a was markedly upregulated in carotid plaques compared with normal carotid artery (Figure 3 B). In vitro experiments in HACECs pretreated with TGF-β1 (5 ng/ml) for 7 days demonstrated a significant increase in miR-126a expression in a time-dependent manner (Figure 3 C). We then performed an in silico search within the sequence of miR-126a for the putative binding site of SENCR, finding a predicted binding motif in miR-126a for SENCR. Subsequent luciferase reporter experiments were applied to confirm the predicted binding site. As shown in Figure 3 D, co-transfection of SENCR luciferase vectors with miR-126a mimics in HACECs for 24 h led to substantially decreased SENCR luciferase activity, while there was no significant change of SENCR luciferase activity with scramble mimics transfection. Altogether, these data suggested that SENCR functioned through direct targeting of miR-126a.
Figure 3
Inhibitory effect of SENCR on miR-126a in ECs. A – Quantitative RT-PCR analysis of eight candidate miRNAs in SENCR siRNA (siSENCR) groups normalized to HACECs transfected with control siRNA (siCon). B – Quantitative RT-PCR analysis of expression of miR-126a in carotid plaques (n = 5) and normal carotid arteries (n = 3). C – Quantitative RT-PCR analysis of expression of miR-126a in response to TGF-β1 stimulation (5 ng/ml). D – Alignment of predicted miR-126a binding sties in the fragment of lncRNA SENCR in Homo sapiens (upper lane). Luciferase activity of HEK293T cells transfected with psi-CHECK2-SENCR luciferase vectors plus miR-126a mimics or scramble mimics was normalized to Renilla activity (lower lane). Experiments were performed three times. Bars indicate mean ± SD
MiR-126 inhibited SMURF2 via directly targeting its 3′UTR
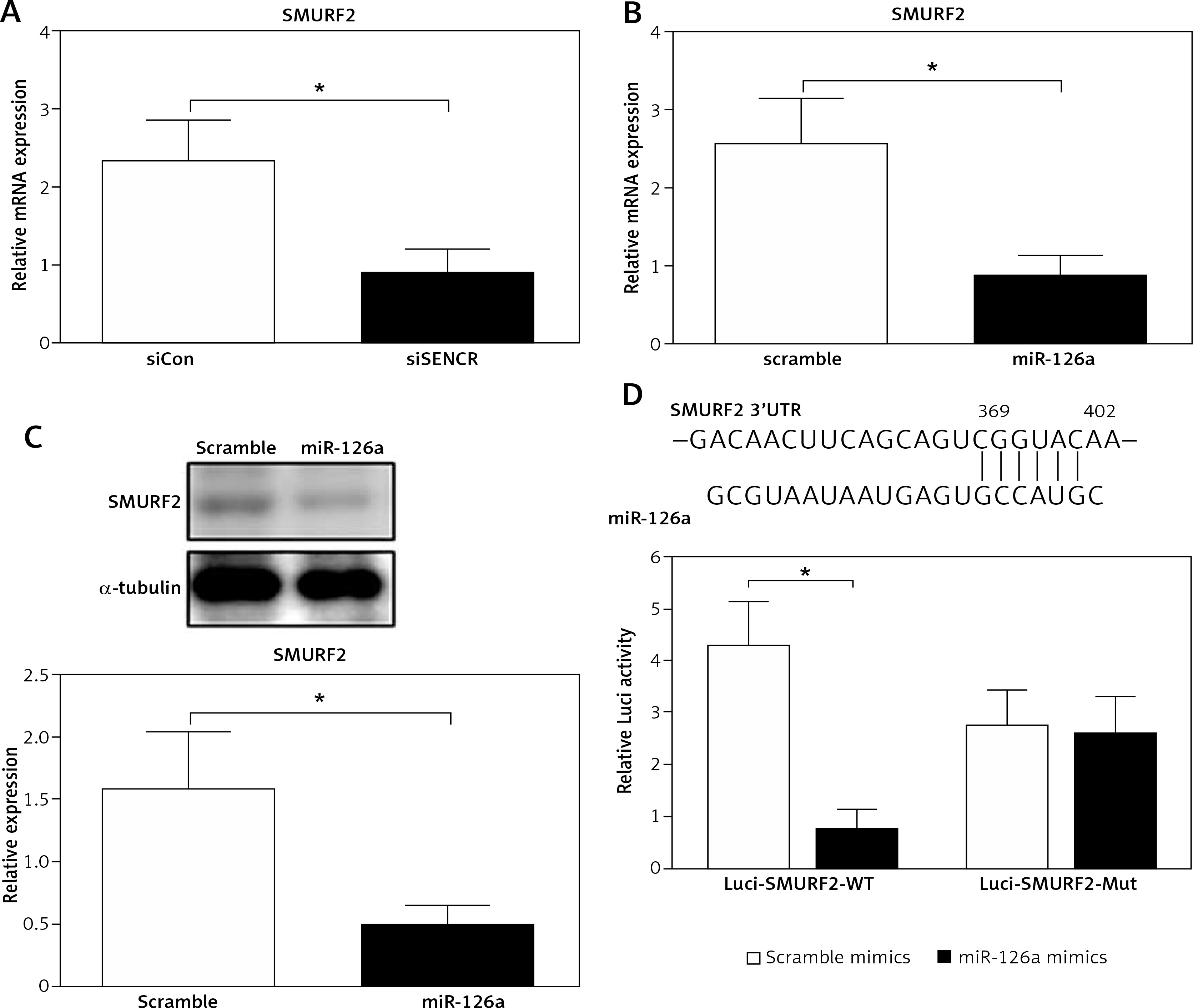
The online in silico miRNA target identification tool TargetScan identified SMURF2 as a potential target of miR-126a. Using quantitative RT-PCR, we found that the mRNA expression levels of SMURF2 were significantly repressed after respective transfection with siSENCR or miR-126a mimics as compared to the control group (Figures 4 A, B). Furthermore, western blots exhibited remarkable inhibition in SMURF2 expression after siSENCR or miR-126a mimics transfection in HACECs as well (Figure 4 C). To confirm the regulatory interaction, the effect of miR-126a on SMURF2 reporter genes was evaluated. Dual-Luciferase Reporter assays elucidated that overexpression of miR-126a significantly suppressed the luciferase activity of SMURF2 3′UTR (Figure 4 D). These results imply that miR-126a directly binds to SMURF2 3′UTR and negatively regulates its expression.
Figure 4
miR-126a directly targets SMURF2. A – Quantitative RT-PCR analysis of expression of SMURF2 after control or SENCR siRNA transfection in HACECs. B – Quantitative RT-PCR analysis of expression of SMURF2 after scramble or miR-126a mimics transfection in HACECs. C – Western blotting of SMURF2 and α-tubulin in HACECs transfected with scramble or miR-126a mimics. D – Construction of the SMURF2 3’UTR luciferase reporters (Luci-SMURF2-WT) and the mutant luciferase reporters (Luci-SMURF2-Mut). Luciferase activity of HEK293T cells transfected with Luci-SMURF2-WT or Luci-SMURF2-Mut vectors plus miR-126a mimics or scramble mimics was normalized to Renilla activity. Experiments were performed in three times. Bars indicate mean ± SD
SENCR-miR126a exerted an inhibitory effect on EndMT via targeting SMURF2
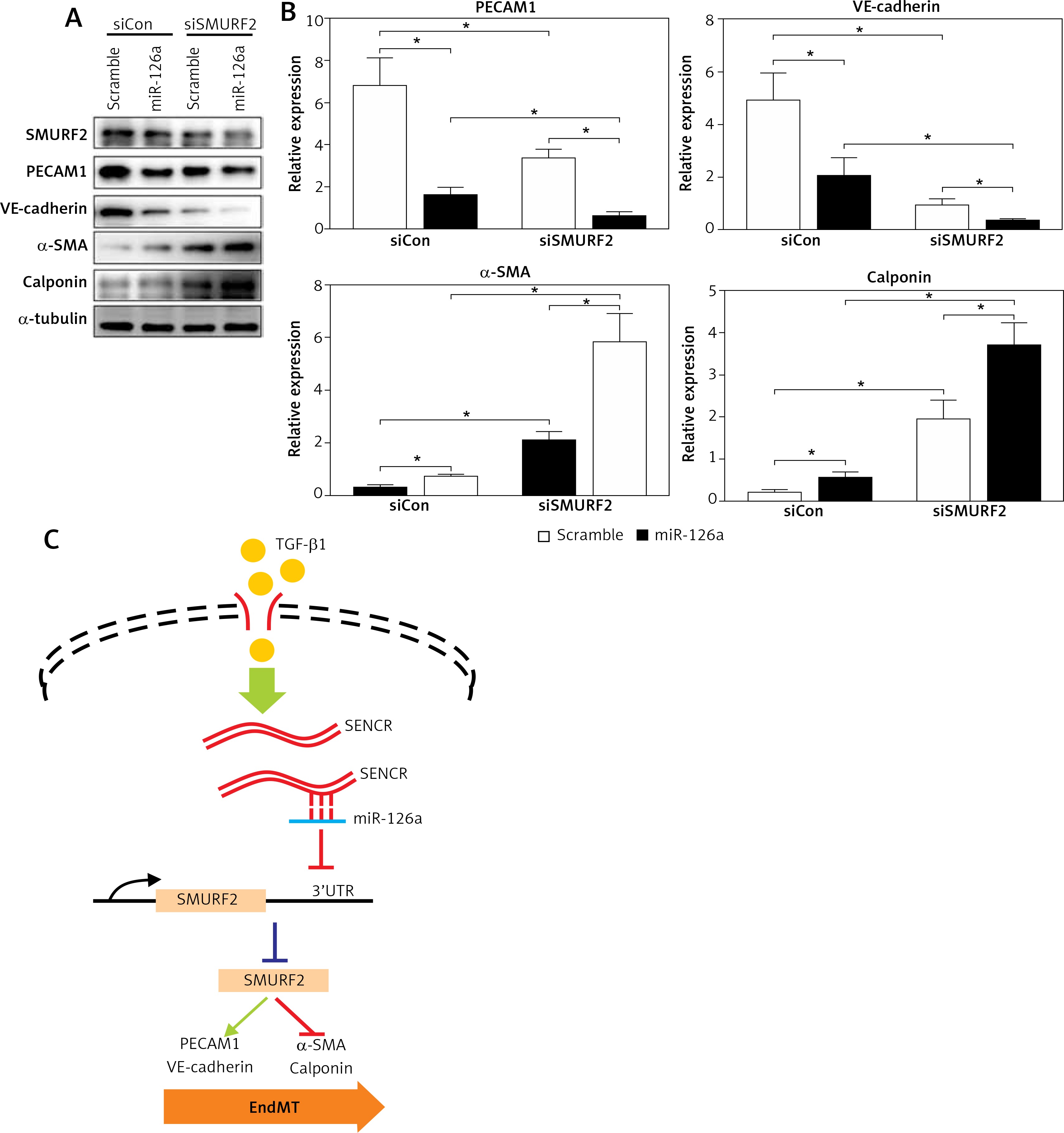
It is known that activation of SMURF2 contributes to the inhibition of TGF-β/Smad signaling. We then examined the effect of miR-126a on smooth muscle markers and endothelial markers in the absence of SMURF2 in HACECs. Consistent with previous reports, knockdown of SMURF2 reduced PECAM1 and VE-cadherin expression and drove α-SMA and calponin expression in HACECs. As shown in Figure 5A and 5B, overexpression of miR-126a moderately restored the elevated expression of the smooth muscle markers α-SMA and calponin under the condition of SMURF2 depletion. Similarly, transfection with miR-126a led to excessive expression of the endothelial markers PECAM1 and VE-cadherin induced by knockdown of SMURF2 in HACECs (Figures 5 A, B). These results suggest that the effect of miR-126a on EndMT progression, at least in part, relies on the inhibitory manner of SMURF2 in HACECs (Figure 5 C).
Figure 5
Effect of miR-126a on EndMT progression is SMURF2 dependent. A, B – Western blotting and quantitative analysis of α-SMA, calponin, PECAM1, VE-cadherin and α-tubulin in HACECs co-transfected with miR-126a mimics and SMURF2 siRNA in the presence of TGF-β1 (5 ng/ml). Experiments were performed three times. Bars indicate mean ± SD. *P < 0.05. C – Schematic diagram of SENCRmiR-126-SMURF2 axis in EndMT progression
Discussion
In the present study, we observed that lncRNA SENCR is decreased in human carotid plaques and depletion of SENCR contributes to the EndMT process in ECs. We provide a mechanistic model demonstrating that SENCR could directly bind to miR-126a and in turn suppress miR-126a expression. Next, miR-126a directly binds to the 3′UTR of SMURF2 and inhibits its expression. Considering the negative role of SMURF2 in TGF-β/Smad signaling, the observation that SENCR-miR-126a mitigates the acquisition of a mesenchymal phenotype of ECs could be explained by the interaction of miR-126a with downstream protein SENCR and the effect of inhibition of SMURF2 on TGF-β/Smad signaling.
Accumulating studies have shown that lncRNA and miRNA expression is frequently tissue- and/or cell-specific with functional specificity in different tissues and cell types as well. LncRNA SENCR is predominantly located in VSMCs and ECs [14]. As a vascular-enriched lncRNA, SENCR overlaps Friend Leukemia Integration virus 1 (FLI1), which is referred to as an early regulator of hemato-endothelial development and a principle regulator of vascular hemostasis [17]. Using high-throughput RNA sequencing to access the changes in the transcriptome of VSMCs, Bell et al. [14] found that knockdown of SENCR altered VSMC phenotype presented as reduction in several VSMC contractile genes’ expression and accompanied by elevation of pro-migratory gene expression. Although SENCR did not control the endothelial differentiation from pluripotent cells, overexpression of SENCR induced the proliferation and angiogenesis of human umbilical endothelial cells [18]. Similar to these results, our data imply that lncRNA SENCR is significantly decreased in carotid plaque compared with normal carotids. Consistent with the observation in carotid samples, SENCR expression was significantly reduced in HACECs after the stimulation of TGF-β, which is considered as a potent atherogenic stimulus.
Our research also shows that loss of SENCR prominently boosts EndMT progression. EndMT is an important process in pulmonary artery remodeling, neointimal formation, atherosclerosis and cardiac hypertrophy [19, 20]. The biological process is complex and is regulated by a complex orchestration of signal pathways [6]. A growing body of evidence has shown that EndMT is primarily governed by common pathways including TGF-β signaling, Notch signaling and Wnt signaling [21, 22]. Consistent with our in vitro observations, two research groups found that TGF-β/Smad signaling promoted Snail-mediated EndMT via Smad-independent signaling and Smad2/3-Slug signaling [8, 23]. In vivo studies using experimental murine models confirmed that TGF-β/Smad-mediated EndMT participated in cardiac fibrosis, atherosclerosis and vein graft remodeling. Conversely, therapeutic strategies with TGF-β neutralizing antibody or some small-molecule inhibitors have been proven to retard the EndMT process and prevent vascular remodeling [8].
Apart from aforementioned traditional pathways, based on miRNA target-prediction bioinformatic analysis, a growing number of miRNAs have been recognized and the target downstream elements were concentrated in the TGF-β/Smad signaling and contribute to the EndMT process [7, 24]. To our best knowledge, one of the first indications of the engagement of miRNAs in EndMT came from Ghosh et al. [25], who observed the differential expression of a specific set of miRNAs during transition of mouse cardiac ECs to a mesenchymal phenotype. Since the EndMT process is crucial for the regulation of skin wound healing, Miscianinov et al. [26] demonstrated that miR-148b accelerated angiogenesis in a mouse model of wound healing and eventually exacerbated skin wound healing in vivo. In vitro mechanistic experiments revealed that overexpression of miR-148b targeted TGFB2 and Smad2, proceeding with recovery of TNF-α/IL-1β-mediated EndMT. Our study identified miR-126a as a direct target of SENCR and promoter of TGF-β-induced EndMT progression. In this regard, Xu et al. [27] found that miR-126a was upregulated by hypoxia during pulmonary hypertension and contributed to EndMT through the p85-β/p-AKT pathway.
Last, in our study, we identify miR-126a as a novel factor that post-transcriptionally regulated SMURF2 via interaction with SMURF2 3′UTR. SMURF2 is an E3 ubiquitin ligase that functions as an inhibitor of TGF-β/Smad signaling and is involved in a wide variety of cellular responses. E3 ubiquitin ligases play a central role in ubiquitination through catalyzation of the attachment of ubiquitin moieties to target proteins and accelerate the process of protein degradation. It is reported that SMURF2 interplays with TGF-β-responsive phosphorylated Smads and triggers their rapid ubiquitination and degradation in a timely manner [28]. On account of these molecular mechanisms, SMURF2 has been implicated in diverse cellular behavior, including cell polarity, cell invasion and cell migration [29, 30]. However, the exact function of SMURF2 in TGF-β-induced EndMT in HACECs remained poorly understood. On the other hand, scientists focusing on the upstream mechanisms of SMURF2 found that microRNAs regulated SMURF2 and TGF-β/Smad signaling via an epigenetic pattern. For example, both miR-322 and miR-503 bound to the 3′UTR fragments of SMURF2 mRNA and abrogated SMURF2 translation but did not affect total SMURF2 mRNA expression [31]. It is therefore accepted that the SENCR-miR-126a complex is capable of involvement in TGF-β/Smad signaling via interaction with the negative regulator SMURF2. Our present study provides additional evidence that SENCR-miR-126a-mediated SMURF2 activation increases the susceptibility of EndMT under exposure to TGF-β1.
In conclusion, we report for the first time that lncRNA SENCR reduces TGF-β-induced EndMT and sponges miR-126a expression via direct inhibition of the negative regulator of TGF-β/Smad signaling SMURF2.