
INTRODUCTION
Mastocytosis represents a heterogeneous group of disorders characterized by abnormal proliferation and accumulation of mast cells (MCs) in various tissues, including the bone marrow, skin, gastrointestinal tract, and parenchymal organs. The estimated prevalence is approximately 10 cases per 100,000 individuals; however, due to diagnostic challenges, the true prevalence may be underestimated. Clinical manifestations result from the release of mast cell mediators such as histamine, heparin, tryptase, chymase, thromboxane, prostaglandins, leukotrienes, and numerous cytokines and chemokines. These mediators may cause symptoms including pruritus, flushing, hypotension, abdominal pain, diarrhea, syncope, dizziness, headaches, tachycardia, and anaphylactic reactions [1–6]. The general classification of mastocytosis is presented in table 1 [1, 5].
Table 1
Cutaneous mastocytosis (CM) is characterized by the accumulation of mast cells in the dermis. It most commonly affects infants and children. Three main clinical forms of CM are distinguished: maculopapular cutaneous mastocytosis (MPCM), diffuse cutaneous mastocytosis and cutaneous mastocytoma [1, 4, 5]. The diagnosis of CM is based on specific clinical and histopathological criteria. The major clinical criterion is the presence of Darier’s sign, defined as the development of erythema and wheals after mechanical irritation of the lesions [4]. Minor criteria include the presence of the KIT D816V mutation in the affected skin and increased mast cell infiltration in histopathological examination (> 15 mast cells in focal aggregates or > 20 mast cells per high-power field at ×40 magnification) [1, 4, 7]. Diagnostic criteria for CM and systemic mastocytosis (SM) are summarized in table 2 [1–3, 5].
Table 2
Within MPCM, two variants are recognized: monomorphic and polymorphic. The monomorphic variant is characterized by small, round, brown or reddish lesions typically measuring a few millimeters in diameter. The polymorphic variant presents with lesions of varying sizes (from several millimeters to several centimeters), which may be brown, red, or yellowish in color [1, 4, 5]. Some adult patients present with telangiectatic macular lesions, most commonly located on the chest, neck, and upper back. These findings are characteristic of telangiectasia macularis eruptiva perstans (TMEP), which was previously considered a separate subtype of CM but is currently classified as a variant of MPCM [4]. Clinical forms of CM and their characteristic features are presented in table 3 [1, 4–6].
Table 3
Adult patients usually present with indolent systemic mastocytosis (ISM), which is the most frequent form of SM and is often associated with cutaneous manifestations [4]. SM may present with symptoms involving multiple organ systems, including weight loss, cardiovascular manifestations (arrhythmias, ischemic heart disease), hematologic abnormalities, skeletal involvement (osteoporosis, osteolysis, bone pain), gastrointestinal symptoms (abdominal pain, peptic ulcer disease, oral ulcers, malabsorption, intestinal motility disorders), neurological manifestations (headaches, depression, fatigue), and hepatosplenic involvement (hepatomegaly, liver dysfunction, ascites, portal hypertension, splenomegaly, thrombocytopenia) [1, 8, 9].
Antihistamines play a key role in the management of symptoms in patients with CM. In cases of severe pruritus, disodium cromoglycate may be beneficial. Patients with gastrointestinal symptoms may require H1 and H2 receptor antagonists and proton pump inhibitors. Additional therapeutic options include psoralen ultraviolet A (PUVA) therapy and topical glucocorticosteroids. Due to the risk of anaphylaxis, patients should be provided with an emergency kit containing epinephrine and glucocorticosteroids [1].
OBJECTIVE
This study aims to present a case of a patient diagnosed with MPCM, with particular emphasis on the disease course, exacerbating factors, and therapeutic approach.
CASE REPORT
A 45-year-old woman with a several-year history of skin lesions was admitted to the Dermatology Clinic for diagnostic evaluation and treatment. Exacerbation of the lesions was observed after exposure to various contact and inhalant allergens, including grasses, tree pollen, cat allergens, and food allergens such as celery, carrot, hazelnuts, apple, and plums. The patient had a history of both food and inhalant allergies and had undergone specific immunotherapy 2 years earlier. Family history was negative for dermatological diseases. The patient was under periodic follow-up at the Gynecologic Oncology Clinic due to uterine fibroids and ovarian cysts.
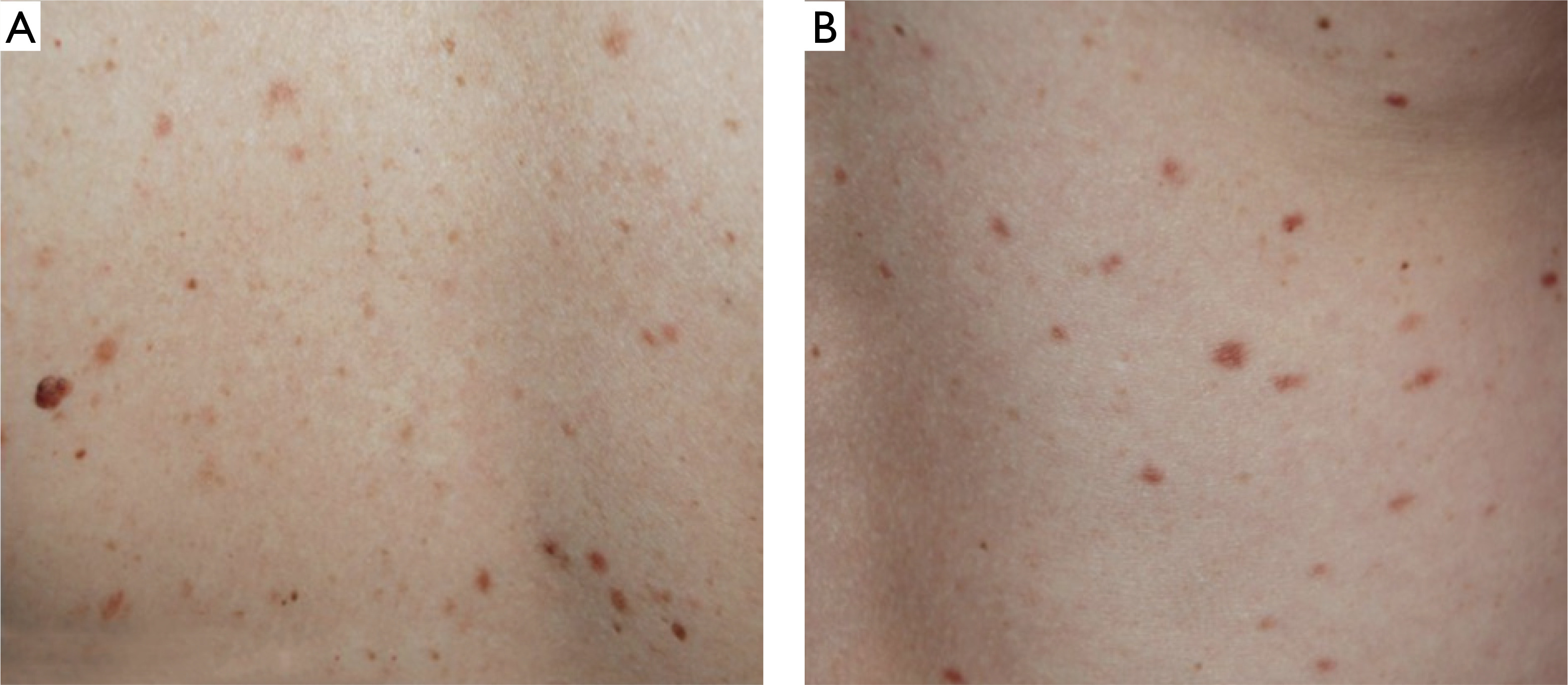
During admission, maculopapular lesions with uneven pigmentation, red-brown in color, were observed on the chest and back (figs. 1 A, B), and to a lesser extent, on the proximal parts of the upper and lower limbs. Darier’s sign was negative (figs. 2 A, B). The patient did not report pruritus and denied associated symptoms such as fever, weakness, syncope, weight loss, diarrhea, or other systemic manifestations.
Figure 1
Clinical presentation at admission: (A) brown-red, round lesions on the trunk and extremities and (B) maculopapular, red-brown lesions on the back and extremities
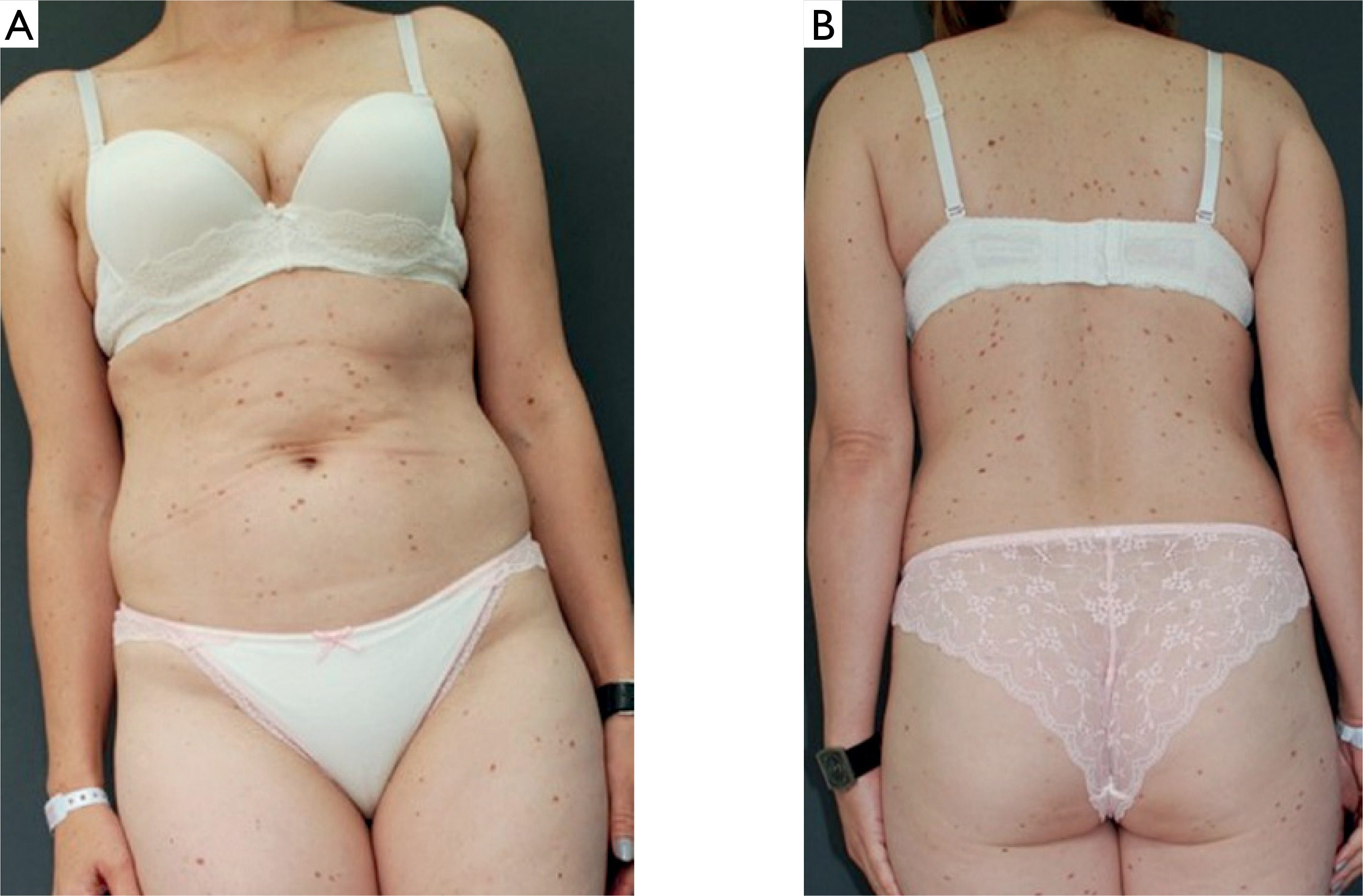
Figure 2
A, B – Clinical presentation at admission: maculopapular lesions with negative Darier’s sign
Laboratory tests revealed an elevated serum tryptase level (16.7 ng/ml; normal: 5.0–11.0 ng/ml). All other laboratory parameters were within normal limits. Histopathological examination of the skin biopsy showed superficial perivascular multifocal infiltration of dispersed mast cells accompanying by single eosinophils around dilated superficial vascular plexus in the dermis. Based on these findings, the pathologist diagnosed TMEP. In previous classifications, TMEP was considered a distinct subtype of CM; however, it is currently regarded as a variant of MPCM.
Treatment included oral bilastine (20 mg once daily), oral prednisone (30 mg/day for 10 days, gradually tapering), and topical mometasone ointment. A significant improvement in cutaneous symptoms was observed. Additional examinations, including abdominal ultrasonography, chest radiography, and bone marrow biopsy were performed to exclude SM.
After discharge, the patient was advised to continue chronic bilastine therapy to control symptoms. She was also educated about the use of epinephrine in the event of an anaphylactic reaction and advised to avoid histamine-releasing factors such as alcohol, aspirin, opioids, heat, excessive physical exertion, anticholinergic drugs, and allergens to which she is sensitive. Further follow-up to evaluate SM was recommended.
DISCUSSION
The diagnosis of MPCM is based on clinical presentation, laboratory findings, and histopathological examination. Early recognition of the disease is crucial to reduce the risk of anaphylaxis and to monitor potential progression to SM. Appropriate management is essential to improve disease control and maintain a satisfactory quality of life in patients with CM. However, establishing the diagnosis may be challenging due to the rarity of the condition and its often non-specific clinical presentation. The diagnostic process includes detailed medical history taking, through clinical evaluation, and appropriate laboratory and histopathological investigations. Management should also include patient education regarding the disease course, potential exacerbating factors, and therapeutic strategies.
Monitoring of CM primarily involves regular clinical assessment with particular attention to cutaneous and systemic symptoms. Measurement of serum tryptase levels (reference range: 5.0–11.0 ng/ml), complete blood counts, and basic biochemical tests are also recommended. Additional examinations may include abdominal ultrasonography. Evaluation of other organs may be required to detect early signs of systemic involvement. Bone mineral density assessment using dual-energy X-ray absorptiometry (DEXA) may be indicated because of the increased risk of osteoporosis and osteopenia. The frequency of monitoring depends on the patient’s clinical status and laboratory findings. An increase in serum tryptase levels or abnormalities in blood counts may indicate disease progression and should prompt further diagnostic evaluation [2, 10]. Regardless of serum tryptase concentrations, patients with confirmed CM should undergo regular clinical follow-up and extended diagnostic evaluation for SM. Referral to a hematologist or allergologist is recommended for specialized assessment and risk stratification. Long-term outpatient follow-up is necessary to enable early detection of potential progression to SM.
Patient education is a key component of disease management and should include information about factors that may exacerbate symptoms or trigger anaphylactic reactions. These include pain, sudden temperature changes (e.g., during bathing), physical exertion, emotional stress, infections, fever, sun exposure, invasive medical procedures, mechanical irritation of skin lesions, Hymenoptera stings, certain foods and food additives, alcohol, and selected medications, including local and general anesthetics and iodine contrast agents [11, 12]. Appropriate prophylaxis and preparation before invasive procedures significantly reduce the risk of anaphylactic reactions. Patients should be equipped with an emergency kit containing epinephrine and trained how to use it. Education of family members or close contacts regarding the management of anaphylaxis is also recommended. Additional preventive measures include medical identification bracelets or information cards for emergency situations [2, 3, 11–16].
Treatment of CM focuses primarily on the control of mediator-related symptoms. Antihistamines targeting H1 and H2 receptors constitute the cornerstone of therapy. Although glucocorticosteroids are not considered first-line treatment, they may be useful during severe cutaneous exacerbations or serious allergic reactions. Additional therapeutic options include leukotriene receptor antagonists, sodium cromoglycate, and, in selected cases, non-steroidal anti-inflammatory drugs (NSAIDs), provided that hypersensitivity to these agents is excluded. Topical glucocorticosteroids and phototherapy, including PUVA, ultraviolet A1 and B, may help alleviate skin symptoms associated with mediator release. Because mastocytosis shares certain immunological features with allergic disorders, treatment strategies are often based on available clinical case reports. Therapeutic options for CM and SM are summarized in table 4 [2, 8, 11, 13, 16–24].
Table 4
CONCLUSIONS
Adult patients with CM require multidisciplinary follow-up involving collaboration between dermatologist, hematologist, and allergologist [6].










