Introduction
Malignant tumors, especially the solid ones, are the leading cause of mortality. Surgical resection, chemotherapy and radiotherapy are still the mainstay treatment modalities for solid tumors. The endeavors to improve clinical outcomes turned out to be disappointing. Even with standard treatments, it is very frequent that cancers become resistant with relapses and severe side effects. Numerous factors cause the delayed diagnoses in the large majority of the cancer patients, and optimal treatments are not applicable for those at the advanced stages. For the patients with distal metastasis, the treatments of choice are palliative. The results after analyzing 36 different types of tumors in 185 countries revealed approximately 18.1 million new cancer cases and 9.6 million cancer-related death worldwide in 2018. The top 10 different types of solid tumors account for over 65% of newly diagnosed cases and mortality [1]. The high lethality of solid tumors pose a great threat to patients and has become an enormous burden globally. Therefore, it is imperative to find new and more effective treatments to cure them.
Adoptive cell therapy (ACT) is carried out through ex vivo expansion, modification and transfer of immune cells to a recipient to elicit an anti-cancer effect [2]. A recently-developed ACT approach is to generate tumor-specific T cells by introducing chimeric antigen receptor (CAR) into T cells (CAR-T) [3]. CARs endow T cells enhanced tumor-specific cytotoxicity, capability to disrupt tumor immunosuppressive microenvironment and to survive in cancer patients persistently [4]. The specificity of CAR-T cells rests on a single chain antibody against a tumor specific/associated antigen (TSA/TAA). Thus, careful selection of the antigen targets is critical to successful implementation of the CAR-T cell therapy. Mesothelin (MSLN), a membrane-bound surface glycoprotein expressed by various solid tumors, is emerging as a potential target for mesothelin-expressing malignancies due to its differential overexpression and tumorigenic characteristics in the development and progression of cancers [5, 6].
CAR-T for cancer adoptive cell immunotherapy
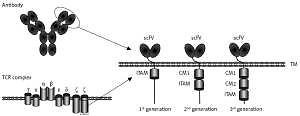
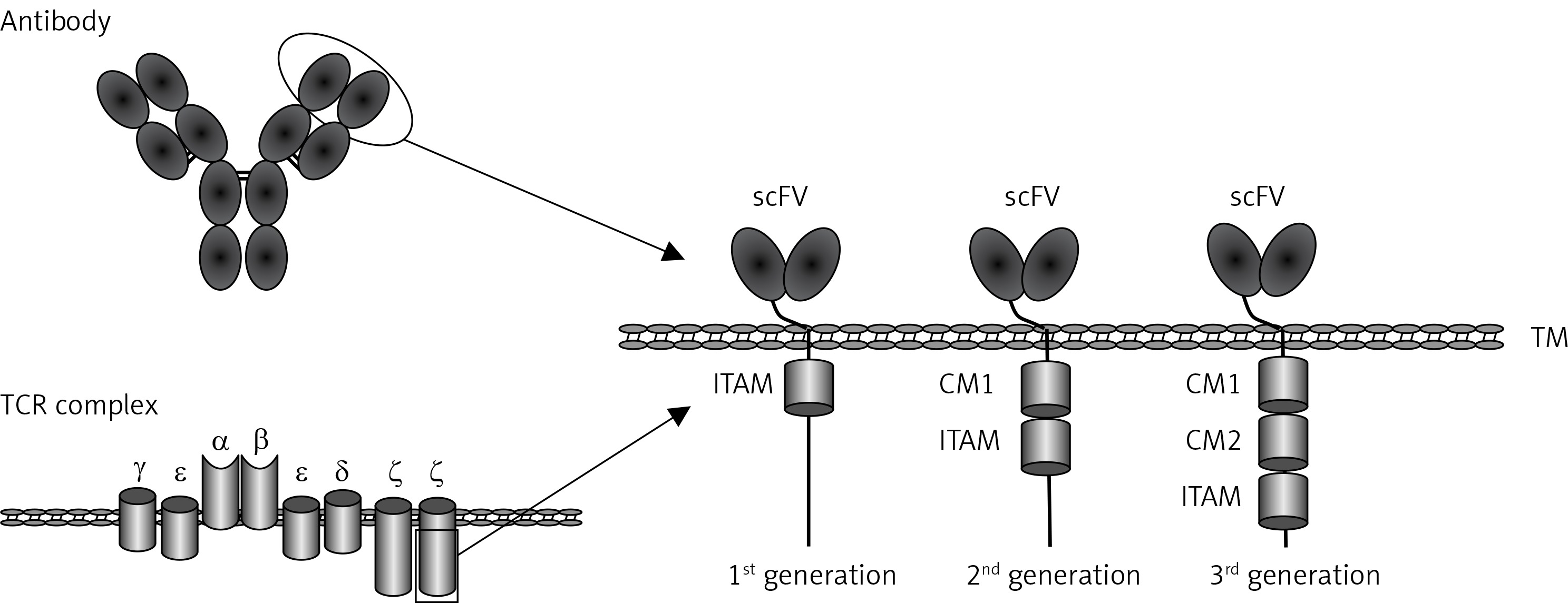
CARs are synthetic receptors able to enhance the anti-tumor efficacy of T cells [7]. A regular CAR consists of an extracellular single-chain variable fragment (scFv) of an antibody to recognize targets, a hinge region to provide flexibility for the scFv, a transmembrane region, intracellular co-stimulatory molecules and CD3ζ chain [8]. Upon specific binding of the scFv to TSAs/TAAs, the signaling domains stimulate T-cell proliferation, cytolysis and cytokine secretion, eventually eradicating target tumor cells (Figure 1).
Figure 1
Schematic structure of chimeric antigen receptor (CAR). CAR consists of a single chain variable fragment (scFv), the hinge and transmembrane (TM) region, co-stimulatory molecules (CM) and human CD3z chain
TCR complex – T cell receptor complex, ITAM – immunoreceptor tyrosine-based activation motif.
CAR-T cells have demonstrated several advantages in the context of adoptive cell therapy [3, 5]. Firstly, CARs recognize cell surface molecules in a major histocompatibility complex (MHC)-independent manner without compromising the specificity and magnitude of T cell response, which helps maintain anti-cancer efficacy even if the expression of MHC is down-regulated in malignancies. Secondly, CAR can recognize a wide array of antigens expressed on the surface of tumor cells including proteins, carbohydrates and gangliosides. Thirdly, the modification of intracellular signaling domains may compensate the down-regulation of co-stimulatory molecules induced by tumor cells. Incorporation of costimulatory molecules such as CD28 and 4-1BB significantly enhances T-cell expansion, survival, cytokine secretion and tumor lysis. Lastly, because of their ability to expand ex vivo rapidly, CAR-T cells may become the primary candidate for future “off-the-shelf” therapy.
CAR-T cell therapy has demonstrated satisfactory efficacy for hematological malignancies including acute lymphocytic leukemia (ALL), chronic lymphocytic leukemia (CLL), and lymphoma in multiple centers worldwide [9]. A meta-analysis of the efficacy of anti-CD19/CD20 CAR-T cells in relapsed or refractory B-cell malignancies demonstrated that the rate of the pooled response, 6-month and 1-year progression-free survival (PFS) were 67%, 65.62% and 44.18%, respectively [10]. Another multicenter study including 391 patients with anti-CD19 CAR-T demonstrated that the pooled rate of complete response and partial response were 55% and 25%, respectively [11]. In accordance with findings, a systemic review of 16 studies including 195 patients with B-cell malignancies showed an overall response rate of 61% with complete response being 42% and partial response being 19% [12]. These results are encouraging with regards to the treatment of relapsed/refractory B lymphoma and leukemia, especially in ALL patients. Better results have been reported by several groups showing a response rate of over 80% in patients with relapsed and refractory B-ALL receiving anti-CD19 CAR-T cells [13, 14].
Despite this success, CAR-T cell therapy does have some limitations. The recognition of CAR-T cells solely depends on the cell surface antigen, while T cell receptor (TCR) is able to interrogate the intracellular proteome of tumor cells. In addition, adverse effects have been reported in different clinical studies with CAR-T cell therapy such as on-target/off-tumor toxicities, cytokine release syndrome (CRS), tumor lysis syndrome (TLS), macrophage activation syndrome (MAS), neurological damage, etc. More comprehensive studies are warranted to tackle these safety issues, possibly by selecting optimal target antigens, controlling the cells through integrating gene switch, etc. [15, 16]. Furthermore, the results of the CAR-T cell therapy are disappointing because of the inherent features of solid tumors.
Challenges for CAR-T of solid tumors
The success in the treatment of hematological malignancies raises a great interest to utilize CAR-T cells to treat solid tumors with CAR-T cells. However, several obstacles must be tackled before its successful use [16, 17]. The heterogeneity of solid tumors is an important factor limiting CAR-T cell immunotherapy. It has been noticed spatially and temporally at the genetic, transcriptomic, proteomic and cellular levels [18, 19]. Spatial heterogeneity can be detected in a single tumor at the primary and metastatic sites. In addition, it can also be found in different tumors in the same patient. Even worse, cancer cells may decrease or change the expression of surface antigens during treatment and tumor progression, causing the recurrence of cancer with poor prognosis [20]. An improved understanding of tumor heterogeneity in primary and metastatic tumors may help develop a quick and comprehensive approach to identify the tumor phenotypes for efficient and targeted therapies by virtue of longitudinal analyses of the potential tumor evolution through the disease courses at different stages and the changes of biological features of tumors over time [21–23].
Efficient migration of CAR-T cells to tumor sites is pivotal to optimal antitumor efficacy, especially, for solid tumors [16]. Circulating B cells in hematological malignancies help intravenously infused engineered T cells to travel to tumor sites. On the contrary, special physical barriers in solid tumors make such movement more difficult and these include higher intensity of blood vessels, wider gap of vessel walls, extensive vascular leakage, extracellular matrix proteins, etc. [17, 24]. In addition, T cell migration to tumor sites requires integrins, chemokines, and chemokine receptors. However, genetic modification of the cell cultured in vitro may cause the loss of chemokine receptors, hindering accurate localization of CAR-T cells to the tumor tissues [25]. These factors may cause poor trafficking of CAR-T cells to solid tumors. Intensive and extensive studies have been performed to tackle these problems and these include engineering T cells with chemokine receptors (such as CXCR2, CCR2b, CXCR4), incorporation of heparanase to degrade the extracellular matrix (ECM), and regional delivery of CAR-T cells [26–28].
Even after translocation into tumor sites, the CAR-T cells are in a hostile environment with numerous immunosuppressive factors that impair their anti-cancer functions by deactivation of T cells [29]. For example, some intrinsic pathways upregulate the expression of inhibitory molecules such as PD-1, CTLA-4 and TGF-β [30, 31]. In addition, aberrant metabolism is common in solid tumors and may affect the biological behaviors of T cells. Further, T cells are difficult to survive due to the lack of arginine or tryptophan, hypoxia, and the acidified extracellular matrix because of hypoxia. Finally, a dysregulated cytokine profile (e.g. IL-15) in tumor may change the fate of the infiltrated T cells [32]. To complicate the matters, tumor stromal cells also produce molecules such as TGF-β, IL-10, and indoleamine-2,3-dioxygenase, leading to suppression of the effector functions of T cells. Numerous strategies such as additional modification of CAR-T cells and combinatorial approaches have been tested to surmount these barriers and enhance CAR-T cells efficacy. These include engineering CAR-T cells expressing dominant negative PD-1 receptors or anti-PD-1/PD-L1 agents, engineering CAR to target stromal cells, addition of the oncolytic virus or inhibitors of negative regulators [29].
MSLN, a tumor associated antigen in various solid tumors
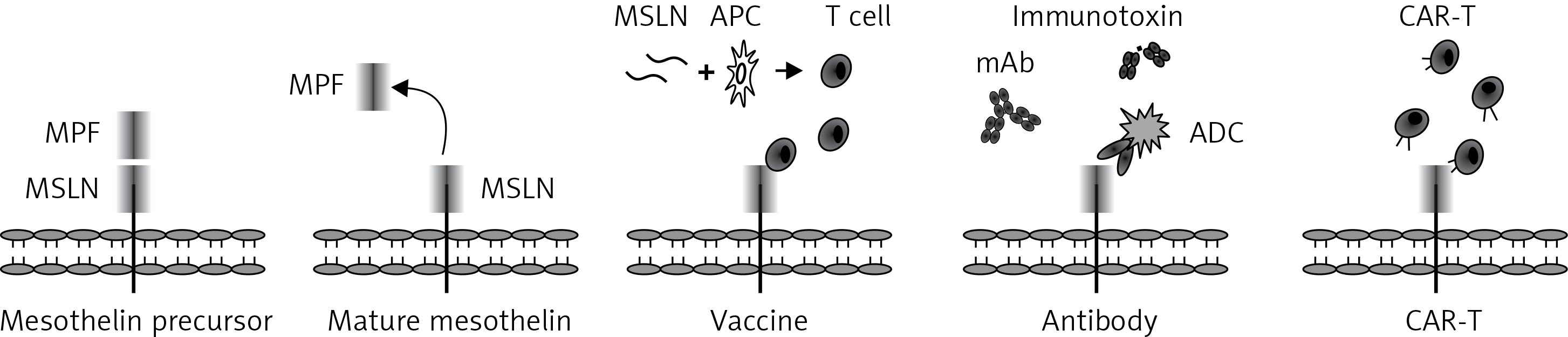
The MSLN gene is located on human chromosome 16p13.3 and composed of 17 exons occupying 8kb of the human genome. Its cDNA is 2138 bp with an open reading frame of 1884 bp, and encodes a 69-kDa precursor protein (628 amino acids) [33, 34]. After cleavage by the furin protease, a 40-kDa C-terminal fragment referred to as MSLN still attaches to the membrane, while a soluble 32-kDa N-terminal fragment is released into the plasma (Figure 2). This soluble molecule is named a megakaryocyte potentiating factor (MPF) due to its capability of inducing megakaryocyte proliferation in vitro [35]. MSLN, a commonly membrane-bound, glycosylphosphatidylinositol (GPI)-linked protein, has 2 variants. Variant-1 with an 8 amino acid insertion is membrane bound, whereas Variant-2 is shed from membrane-bound MSLN and becomes soluble due to the lack of GPI-anchor signal sequence [36]. The soluble form (also known as soluble MSLN-related peptide, SMRP) was initially identified in the plasma of patients with ovarian carcinoma using the OV569 mAb.
Figure 2
Mesothelin structure and strategies of immunotherapy. Mature mesothelin (40 kDa) is attached to the surface of the cell membrane after cleavage of the mesothelin precursor by furin protease, while a soluble 32-kDa N-terminal fragment (megakaryocyte potentiating factor, MPF) is released into the plasma
MSLN – mesothelin, APC – antigen presenting cells, ADC – antibody-drug conjugation, mAb – monoclonal antibody, CAR-T – chimeric antigen receptor-engineered T cells.
The biological function of MSLN remains largely unknown and seems nonessential for normal tissues. MSLN-deficient mice grow and reproduce normally without a detectable phenotype [37]. Nevertheless, it may be involved in cell migration and spreading, the promotion of proliferation, inhibition of apoptosis and chemotherapy resistance in solid tumors. MSLN can act bidirectionally: either interacting with its receptor CA125/MUC16, or directly activating intracellular pathways via its GPI domain. The interaction between mesothelin-CA125 induces a heterotypic cell adhesion resulting in the spreading of malignancies in the body cavities [38]. Overexpression of MSLN enhances proliferation and migration of cells by constitutive activating of nuclear factor κB (NF-κB) and upregulating of MMP-7 and MMP-9 through MAPK pathways [39–41]. Further, MSLN prevents the drug-induced release of pro-apoptotic TNF-α by activating Akt/PI3K pathway, enhances the expression of antiapoptotic genes such as Bcl-2 and Mcl-1, and inhibits the expression of pro-apoptotic factors such as Bad and Bax [42]. However, much needs to be done to unveil the exact function of MSLN in cancer development and progression.
A weak expression of MSLN has been found in the mesothelial cells of the pleura, peritoneum, and pericardium, fallopian tubes, trachea, cornea and tonsils. However, it is overexpressed on various solid tumors including mesotheliomas, pancreatic adenocarcinomas, ovarian cancers, non-small lung cancers, etc. [5]. MSLN is overexpressed in 95% of malignant mesothelioma (MM) patients of the epithelioid form. No overexpression of MSLN is detected in the sarcomatoid form, and the biphasic variant has a low to intermediate MSLN expression depending on the percentage of epithelial components [43]. MSLN is detected in 80-85% of pancreatic ductal adenocarcinoma (PDAC) but not in normal pancreas and chronic pancreatitis [44]. A previous report showed that MSLN was overexpressed in 68.8% of ovarian cancers and 24.2% of borderline-type tumors [45]. The mechanisms underlying overexpression of MSLN in different malignancies is not fully understood. Activation of the Wnt/β-catenin pathway and the methylation of the promoters of certain genes may be involved in the process. Clinical observations have demonstrated that an enhanced MSLN expression and elevated serum SMRP level are associated with progressing tumor burden, increasing stage, and poor overall survival [46, 47]. Tumor cells with an invasive phenotype express high amounts of membranous MSLN. It is overexpressed at lung adenocarcinoma (ADC) and correlates with tumor aggressiveness. Patients with a high MSLN score had reduced OS and recurrence-free survival (RFS) compared with patients with a low MSLN score in multivariate analyses [47].
MSLN as a target in cancer immunotherapy
The high incidence of overexpression and oncogenic characteristic features make MSLN an ideal target for immunotherapy of solid tumors. A variety of strategies targeting MSLN are currently under intensive research and these include as vaccines, a monoclonal antibody, the antibody-drug conjugation, immunotoxin and adoptive cellular therapy (Figure 2).
An MSLN-based vaccine, CRS-207, is a live-attenuated Listeria monocytogenes strain engineered to express human MSLN. After secretion, it incites innate and adaptive immune responses in antigen presenting cells (APC) to target MSLN [48]. Preclinical studies confirmed the ability of such vaccines to elicit an effective antitumor response to MSLN-expressing tumors including PDAC through both T-cell- and antibody-mediated immunity [48, 49]. In addition, CRS-207 was well tolerated in a phase I clinical trial (NCT00585845). Immune induction and activation of MSLN-specific T-cell responses were observed. Among 13 evaluable patients, four achieved stable disease, 37% of patients were alive for more than 15 months [50]. With these data, CRS-207 was combined with GVAX (GM-CSF gene modified tumor vaccine) and low dose cyclophosphamide (Cy) for clinical evaluation. In a phase II clinical trial (NCT01417000), Cy/GVAX combination with CRS-207 improved OS of previously treated metastatic PDAC patients compared to Cy/GVAX alone (6.1 vs. 3.9 months) [51]. Notably, no toxicities were observed in these patients. Besides, a cell-based vaccine (Meso-VAX), in which a secreted MSLN anchored at the cell membrane, elicits antigen specific immunity and potent antitumor effects in mice by combining with IL-12 immunomodulator [52]. MSLN-specific DNA vaccine are also under investigation. CTGF/MSLN DNA with anti-CD40Ab and poly(I:C) vaccinated mice demonstrated robust anti-tumor effects with longer survival and less tumor volumes [53].
Amatuximab (MORAb-009) is a humanized monoclonal antibody (mAb) against MSLN [54]. In preclinical studies, this mAb causes the death of the MSLN expressing cell lines by antibody dependent cell-mediated cytotoxicity (ADCC), and inhibits the interaction between MSLN and CA-125. Two phase I studies were conducted to investigate in vivo distribution and safety of 111In-amatuximab in MSLN-expressing tumors (NCT01521325, NCT01413451), and the dosimetry profile confirmed a significant uptake of the mAb in both primary and metastatic tumors [55]. Another phase I study (NCT00325494) in 24 patients with advanced MSLN-expressing cancers showed a good tolerance with mild drug-related hypersensitivity reaction and dose-dependent immunogenicity [56]. Although no objective partial tumor responses were reported in these studies, stable disease was achieved in a phase I clinical trial (NCT01018784). A subsequent phase II study (NCT00738582) in 89 patients with unresectable malignant pleural mesothelioma (MPM) has been conducted with Amatuximab alone or combination with pemetrexed and cisplatin. Objective response and stable disease were achieved in 40% and 51% of patients, respectively [57].
Anetumab ravtansine (BAY94-9343) is an antibody drug conjugated to DM4 (a tubulin polymerase inhibitor) [58]. When the drug is bound and internalized by MSLN-expressing tumor cells, DM4 metabolites cause cell-cycle arrest and apoptosis. In a phase I trial (NCT02485119), it was well tolerated with an acceptable toxicity profile when intravenously infused every 3 weeks. Another phase I trial (NCT01439152) showed that partial responses (PR) and stable disease (SD) were 19% and 47%, respectively [59]. Currently, some phase I and II trials testing BAY 94-9343 alone or in combination with chemotherapy are being carried out. It has reached phase II as a single agent to treat pancreatic (NCT03023722) and lung cancers (NCT02839681). DMOT4039A is a humanized MSLN mAb conjugated to the antimitotic agent monomethyl auristatin E [60]. A phase I clinical trial (NCT01469793) evaluated DMOT4039A in 40 PDAC and 31 ovarian cancer patients. The dose limiting toxicities (DLTs) were grade 3 hyperglycemia and grade 3 hypophosphatemia in the q3w schedule. Six patients had partial response at 2.4–2.8 mg/kg every 3 weeks. Some other ADCs targeting MSLN such as BMS-986148 and MDX-1204 are under investigation.
SS1P is a recombinant protein drug consisting of the anti-MSLN Fv SS1 fused to PE38, a portion of the Pseudomonas exotoxin A [61]. When internalized to the cytoplasm, it irreversibly modified elongation factor-2 to halt protein synthesis and induced apoptosis. Given its safety and tolerability in phase I monotherapy study, SS1P was combined with pemetrexed and cisplatin for the treatment of chemotherapy-naive patients with unresectable MPM (NCT01445392) [62]. This combinatorial therapy resulted in an objective response rate of 77% without overlapping toxicity. However, the efficacy of SS1P is compromised due to the formation of the antidrug antibody. As such, SS1P is now being administered together with a lymphocyte-depleting conditioning regimen of pentostatin and cyclophosphamide. This combination prevents anti-SS1P antibody formation and demonstrates a remarkable antitumor effect in mesothelioma both in vitro and in vivo. LMB-100 (previously named RG7787) is a de-immunized RIT containing a humanized anti-MSLN Fab fused to PE24 [63]. Given its reduced toxicity and significant antitumor effect in preclinical studies, initial phase I-II studies are recruiting patients with MM (NCT02798536, NCT034367321) and PDAC (NCT02810418). Taken together, these results demonstrate the safety and feasibility of MSLN as a target for CAR-T cells immunotherapy.
MSLN CAR-T cells for solid tumors immunotherapy
To date, significant progresses have been made in the preclinical models using CAR-T cells targeting MSLN (CARTmeso). T cells transduced with anti-MSLN CAR (CD28-4-1BB-CD3ζ) using lentiviral vectors were evaluated for efficacy against MSLN-expressing tumor xenografts in NOD/scid/IL2rγ(–/–) mice [64]. Both intratumoral and intravenous administration of CARTmeso cells resulted in significant shrinkage of tumors. In addition, intratumoral injection of RNA CAR-electroporated (4-1BB-CD3ζ) T cells caused regression of disseminated human mesothelioma xenografts [65]. Further, a fully humanized MSLN-specific P4CAR-T (CD28-CD3ζ) cell was tested in vitro and these cells demonstrated potent cytolytic function when cultured with MSLN-expressing tumor cells at least in part due to the production of proinflammatory cytokines and degranulation. Adoptive transfer of P4CAR-T cells resulted in the regression of large, established tumor in a xenogeneic model of human ovarian cancer [66]. A recent study with fully humanized CARTmeso cells confirmed their ability to kill MSLM-positive pancreatic cells, to penetrate into the tumor, and to significantly suppress the growth of pancreatic cancer PDX engrafted tumor [67].
These promising results from preclinical models support the widespread clinical use of CARTmeso cells for the treatment of the MSLN-expressing solid tumors. Thus far, 25 clinical trials have been registered on the ClinicalTrials.gov, four of which have been completed. In two initial clinical studies, anti-MSLN CAR mRNA (4-1BB-CD3ζ) electroporated T cells demonstrated good tolerance and minimal on-target/off-tumor toxicity [68, 69]. These CARTmeso have shown potent antitumor activity, the capability to infiltrate into solid tumors and to induce humoral epitope spreading. One patient with MPM achieved partial response for 6 months, and 1 patient with pancreatic cancer achieved stable disease (NCT01355965). Stable diseases were found in 2 out of 6 patients with chemotherapy-refractory metastatic PDAC, with PFS time of 3.8 and 5.4 months (NCT01897415). A lentiviral-transduced CARTmeso cells trial was conducted (NCT02159716) in patients with PDAC, MPM and ovarian cancer. The CARTmeso cells were well tolerated and could be detected in the blood for up to 30 days [70]. More trials evaluating CARTmeso cells alone or combinatorial strategies have been performed. In an ongoing study, four pancreatic cancer patients are being treated with T cells transduced with lentiviral anti-MSLN and anti-CD19 CAR (4-1BB-CD3ζ), which are expected to attack the B cells and prevent the antibody response against CARTmeso cells (NCT02465983). Notably, a more active, fully human anti-MSLN CAR is being tested to enhance persistence and efficacy in a phase I trial (NCT03054298). Based on preclinical findings that local administration of CAR-T cells was required for optimal antitumor efficacy [26], one trial using iCasp9M28zCAR targeting MSLN was performed to test the safety of intrapleural administration of CARTmeso cells in patients with pleural malignancies (NCT02412469).
Future directions and conclusion
Prompt translation of CAR-T cells therapy to the clinical practice in CD19+ malignancies has raised intensive attention to adoptive cell therapy and engineered T cells. However, this approach has not gained momentum in solid tumors. Preclinical and clinical studies suggest that MSLN-targeted CAR-T cell therapy is a promising strategy due to its safety profile and potent antitumor efficacy in MSLN-expressing malignancies. Because of the complexity of solid tumors, more sophisticated techniques are warranted to eradicate solid tumors with CARTmeso cells including modifying the CAR to secrete chemokines or cytokines receptors, targeting the immunosuppressive tumor microenvironment such as intervening the immune checkpoint on T cells, incorporating suicide genes to improve the safety, etc. We are optimistic that with intensive research, CARTmeso cells will become a potent and efficient treatment method for solid tumors in foreseeable future.