Introduction
The anti-inflammatory drug minocycline is broadly known for its function in alleviating neurological disorders including spinal cord injury [1–4]. Traumatic spinal cord injury includes a complex pattern of pathophysiological processes out of which mitochondrial activity in cell survival and death holds a greater impact [5]. Minocycline’s efficacy to impede mitochondrial cell death has been established previously [6, 7]. However, the effect of minocycline on mitochondrial activity and hypoxia-inducible factor-1α (HIF-1α) regulation following traumatic spinal cord injury has not been studied.
Minocycline has long been known to be efficient as an anti-inflammatory and anti-microbial drug [8, 9]. The constant research on this drug opened new avenues towards the treatment of several neurological disorders including amyotrophic lateral sclerosis (ALS), multiple sclerosis, Huntington’s disease and ischaemic stroke [10, 11]. However, minocycline treatment worsened ALS in a phase III clinical trial whereas it provided promising results in stroke and multiple sclerosis. This discrepancy could be due to the increased activity of minocycline in the inflammatory response [12]. Minocycline directly targets the mitochondrial proteins and impedes the release of factors responsible for cell death which could either be caspase-dependent (Smac/Diablo, cytochrome c (cyt c)) or caspase independent (apoptosis-inducing factor) [6].
Fas cell surface death receptor (FAS)-mediated apoptosis in spinal cord injury is reported to require mitochondrial signalling and the inhibition of FAS ligand (FASL) protects the cells from undergoing apoptosis [13]. Another important mitochondrial target gene is HIF-1α, and the expression of HIF-1α is time dependent, leading to cell death or cell survival following neuronal injury [14]. Mounting evidence suggests that the stabilization of HIF-1α following neuronal damage protects the hypoxic cells from apoptosis [15, 16]. Interestingly, activation of HIF-1α and FAS converge in the release of pro-apoptotic proteins, but they are independent mechanisms. HIF-1α stabilizes the p53 pathway, whereas FAS mediates cell death by activation of caspase 8 and increase of BH3 interacting domain death agonist (BID). Both mechanisms interrupt the binding between B-cell lymphoma 2 (BCL2) and BCL2 associated X protein (BAX), which leads to the oligomerization of BAX and eventually permeabilization of the mitochondrial membrane [17]. In this study, we hypothesised that minocycline modulates mitochondrial cell death via mechanisms interlinking HIF-1α stabilization and FASL-mediated apoptosis.
Material and methods
Inducing spinal cord injury
Male Sprague Dawley rats (N = 15) weighing approximately 230–250 g were obtained from SLAC company, Songjiang District, Shanghai, China. The study was approved by the local ethical committee and performed according to the National Research Council Guide. Animals were anaesthetised using 50 mg/kg of chloral hydrate and exposed to traumatic spinal cord injury as previously described [18]. Briefly, the T9-T10 vertebral level was subjected to laminectomy without damaging the dura. A force-calibrated weight drop device (originally designed at New York University) was used to induce traumatic injury once the T-8 and T-11 spinous processes were steadily clamped and the dorsal cord surface was put through a weight drop impact according to the procedural guidelines [19]. Following the injury, exposed regions were closed and the animals were placed in a chamber under optimum conditions (temperature of 22 ±0.5°C and humidity of 50 ±10%, pathogen-free environment).
Drug administration
Rats that underwent traumatic spinal cord injury were immediately injected intraperitoneally with 90 mg/kg of minocycline (Sigma-Aldrich, Huangpu, Shanghai, China) and later a 45 mg/kg dose twice a day (every 12 h) (n = 5). The treatment dose was selected based on a previous study where minocycline administration exhibited neuroprotection in brain trauma [10]. The treatment continued until 2 weeks, after which the spinal cord tissue of around 20 mm was obtained from the site of the lesion by anesthetising each rat. Injured animals injected with the sterile phosphate buffered saline (PBS) (n = 5) served as the vehicle (control group) and another group included normal healthy animals (n = 5) with no injury.
TUNEL assay
TUNEL staining was performed using the ApopTag plus peroxidase in situ apoptosis detection kit (Sigma Aldrich, Huangpu, Shanghai, China) according to the manufacturer’s instructions. Before performing the staining, spinal cord sections were processed by fixing in 4% paraformaldehyde overnight and embedded in paraffin.
ELISA assay
Samples were prepared by homogenising the tissue of spinal cord segments and were subjected to ELISA using specific kits that are commercially available (Enzo life sciences, Chang Shou, Shanghai China). The kits specific to Smac (Diablo), cyt-c, HIF-1α, and FASL and TNF-α were purchased and the protocol as described by the manufacturer was followed.
Immunohistochemical staining
Homogenised samples were fixed for 10 min in 4% PFA in PBS followed by incubation with primary antibodies (rabbit anti-FasL dilution 1 : 300, anti-HIF-1α dilution 1 : 500) (Santa Cruz Biotechnology, Chai Wan, Hong Kong, China) overnight at room temperature. After that, the samples were incubated for 2 h at room temperature with goat secondary antibody (1 : 1000) (Santa Cruz Biotechnology, Chai Wan, Hong Kong, China) conjugated with streptavidin peroxidase and counterstained with haematoxylin to detect the staining. The slides were visualised under an Olympus microscope (Olympus, Shinjuku-ku Tokyo, Japan) and images were analysed using Image J software.
qPCR mRNA expression analysis
Homogenised spinal cord tissue segments were used to isolate RNA according to the manufacturer’s instructions (Thermo Fisher Scientific, Pudong, Beijing, China). RNA extracted was quantified using the NanoDrop 1000 system (Thermo Fisher Scientific, Pudong, Beijing, China) and then converted to cDNA using the iScript cDNA synthesis kit (Bio-rad, Haidian District, Beijing, China). Expression of Smac/Diablo, cytochrome c, FASL and HIF-1α and p53 was identified by performing SYBR green (Bio-rad, Haidian District, Beijing, China) labelled qPCR (Applied Biosystems, Chaoyang District, Beijing, China). Primers specific for each gene were obtained commercially (Bio-rad, Haidian District, Beijing, China). β-Actin was used as an internal control to normalize the mean Ct values.
Western blot analysis
Protein was isolated from homogenised samples and quantified using the BCA kit (Sigma-Aldrich, Huangpu, Shanghai, China). Ten percent SDS PAGE was done to separate the proteins and then transferred to a nitrocellulose membrane by means of electroblotting technique. Following this, the membrane was blocked using 5% blocking solution (non-fat milk). The membrane was kept in the blocking buffer containing primary antibodies overnight at 4°C (rabbit anti-cytochrome c 1 : 500, anti-smac 1 : 1000, anti-HIF-1α 1 : 500, anti-p53 1 : 1000, anti-FASL 1 : 300, anti-caspase-8 1 : 1000, anti-BID 1 : 500 and anti-β-actin 1 : 1000 (Santa Cruz Biotechnology, Chai Wan, Hong Kong, China). Next day, goat secondary antibody conjugated with horseradish peroxidase was added in the dilution 1 : 1000 in blocking buffer (non-fat milk). A chemiluminescence kit (Thermo Fisher Scientific, Pudong, Shanghai, China) was used to detect the proteins. The sample used was the mitochondrial fraction isolated from the rat spinal cord, as previously described [20].
Results
Effect of minocycline in inhibiting cell death following spinal cord injury
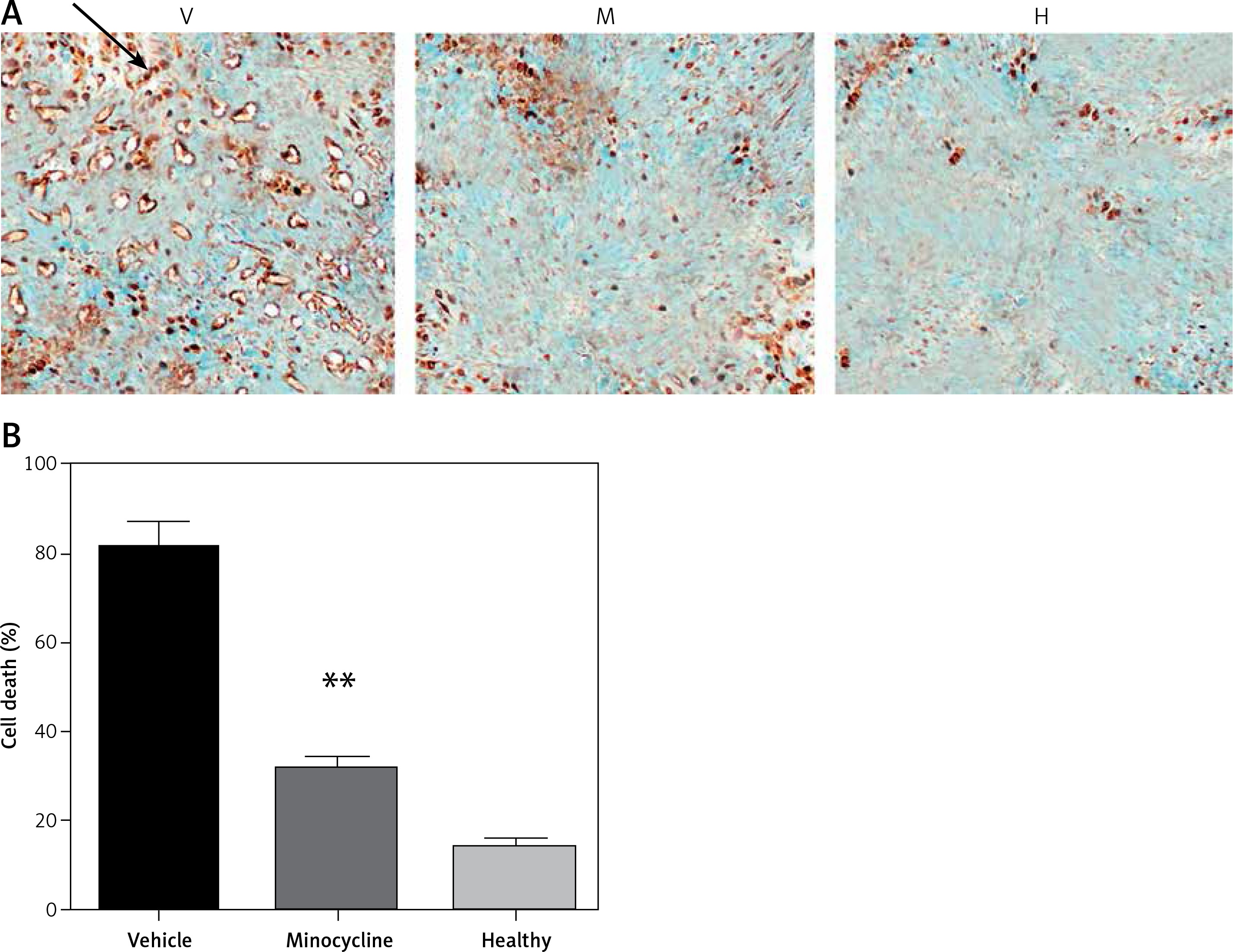
TUNEL staining showed that minocycline had a significant effect in reducing cell death (**p < 0.01 vs. vehicle). The number of TUNEL positive cells in vehicle and minocycline-treated samples was counted. The samples retrieved from the normal rats did not exhibit TUNEL positive cells. The results were in compliance with the previous effects of minocycline on cell death following traumatic spinal cord injury. The effect of minocycline following the injury is shown in Figures 1 A, B.
Figure 1
Evaluating the effect of minocycline on cell death through TUNEL assay after spinal cord injury. A – TUNEL staining shows cell death in vehicle (V), minocycline-treated (M) and healthy groups of animals (H). Reduced cell death was observed in minocycline-treated group (400×). Arrow indicates the positive TUNEL stained cell. B – Cell death was quantified and graphically represented. There was an approximately three-fold reduction in cell death in the minocycline-treated group compared to the vehicle. All values are represented as mean ± SEM and values with p < 0.05 were considered significant
*P < 0.05 vs. vehicle group, **p < 0.01 vs. vehicle group.
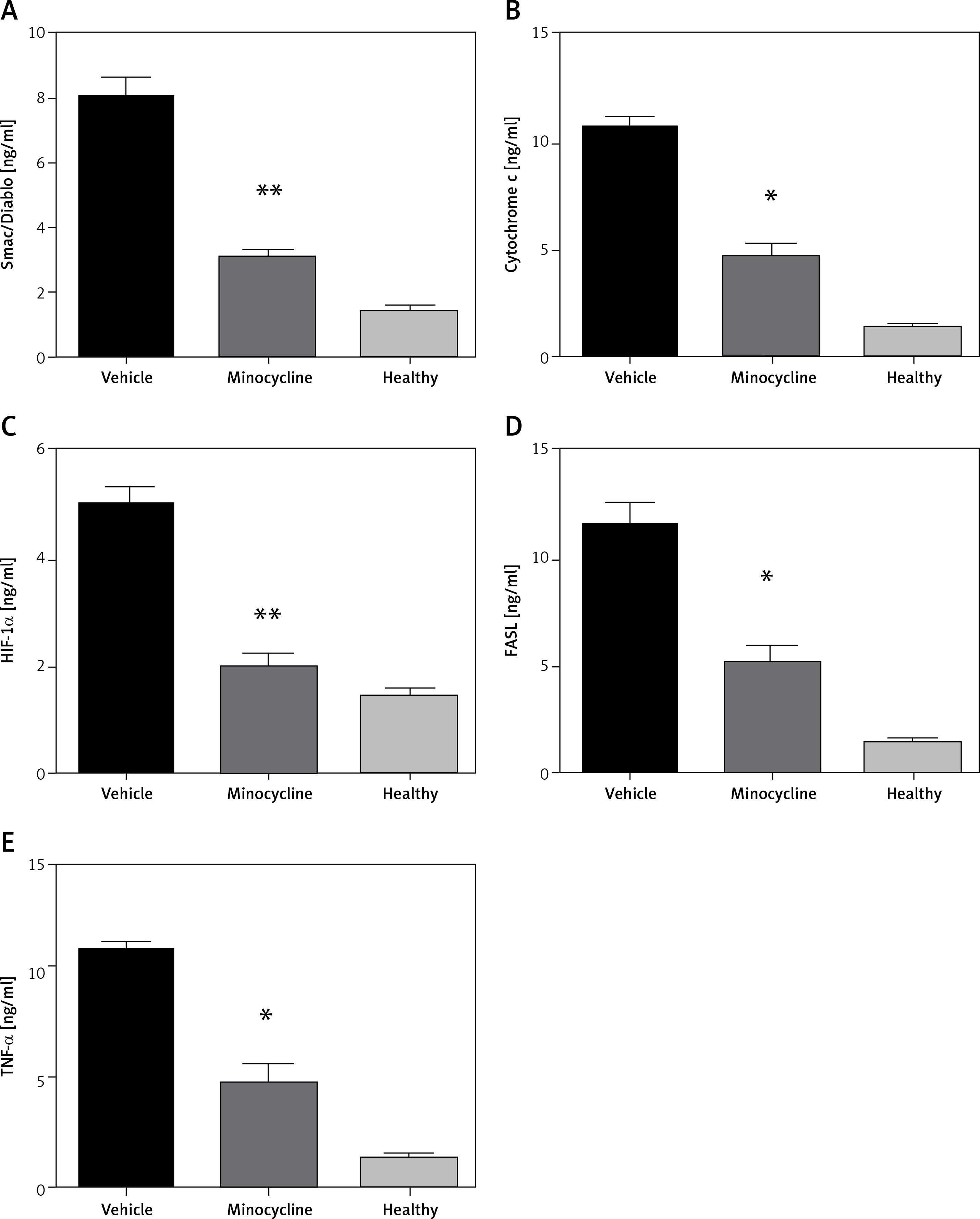
Quantification of Smac (Diablo), cytochrome c, HIF-1α, FASL and TNF-α
ELISA quantified the release of Smac (Diablo), cytochrome c, HIF-1α, FASL and TNF-α to demonstrate activation of the intrinsic and extrinsic apoptotic pathway following spinal cord injury. The increased release of Smac/Diablo, cytochrome-c and FASL indicates greater cell death through caspase-dependent apoptosis [6]. FASL mediates cell death through pro-inflammatory cytokines and the level of TNF-α was quantified to confirm the association of minocycline with FASL [13]. The level of HIF-1α following neuronal injury is highly time-dependent, leading to cell survival. The results demonstrated that there was an increase in the level of mentioned proteins (Smac/Diablo, cytochrome c, HIF-1α, FASL and TNF-α) following the injury and the minocycline treatment modulated the protein content (*p < 0.05 vs. vehicle, **p < 0.01 vs. vehicle) (Figures 2 A–E). The treatment with minocycline reduced the level of mitochondrial proteins (Smac/Diablo and cytochrome c) associated with cell death. The action of minocycline on these mitochondrial proteins was also hypothesised to be related to the alteration in HIF-1α and FASL levels. Figure 2 demonstrates a significant reduction in the release of mitochondrial proteins associated with cell death along with the reduction of HIF-1α.
Figure 2
ELISA quantification of release of mitochondrial proteins. The release of Smac/Diablo,cytochrome c, HIF-1α, FASL, and TNF-α was quantified using ELISA following spinal cord injury from the minocycline-treated group of animals, vehicle group and healthy group (A–E). There was a significant reduction in the release of these proteins compared to the vehicle group. All the values are expressed as mean ± SEM and were considered significant at p < 0.05
*P < 0.05 vs. vehicle group, **p < 0.01 vs. vehicle group.
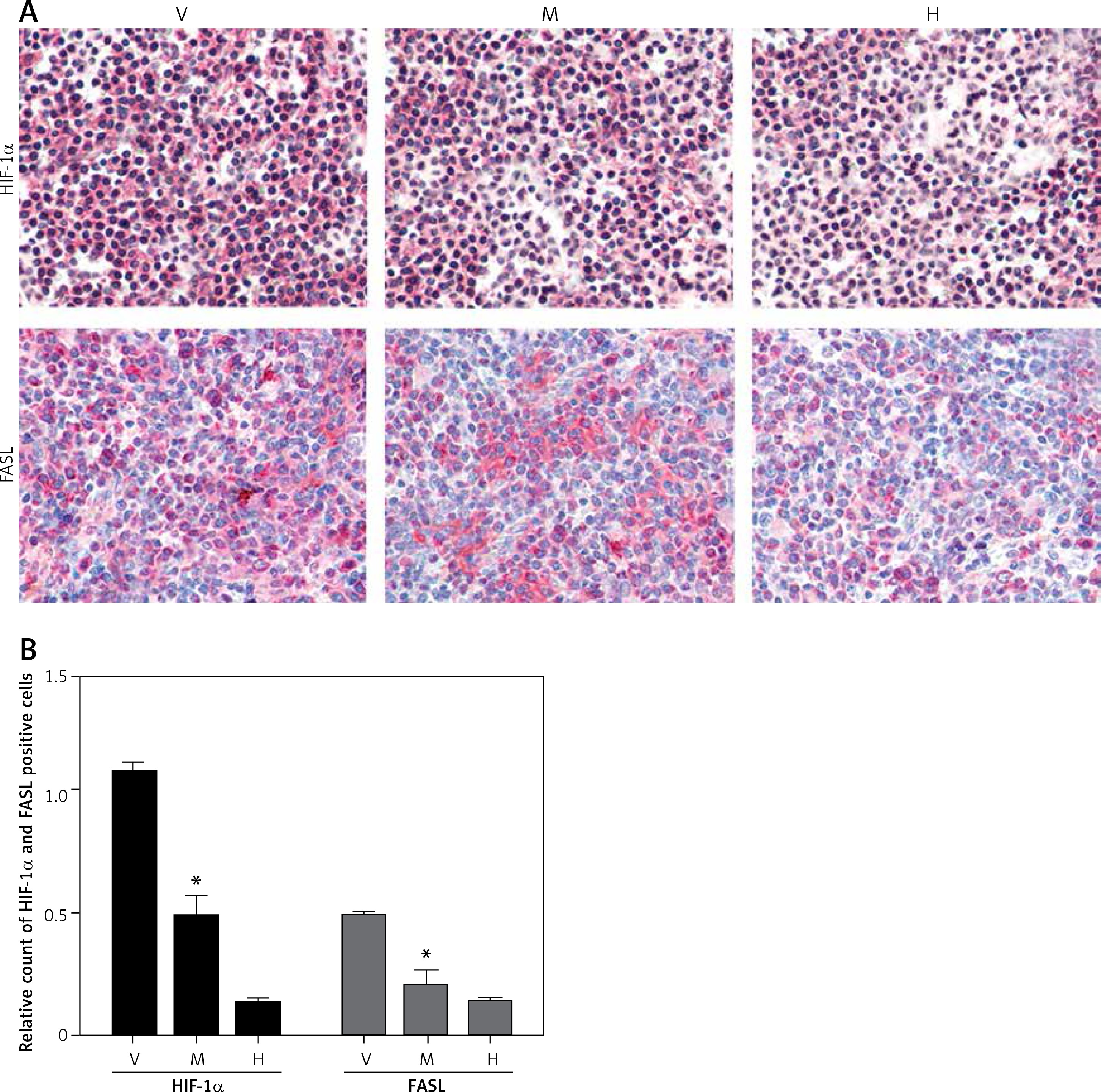
Minocycline treatment reduced the expression of HIF-1α and FASL
Immunohistochemistry from the spinal cord tissue sections (20 mm) from the site of the lesion demonstrated that minocycline reduced the expression of HIF-1α and FASL compared to the vehicle group. The relative count of cells positive for HIF-1α and FASL showed that minocycline significantly reduced the number of cells positive for HIF-1α and FASL. Minocycline treatment might inhibit cells by regulating expression of FASL and HIF-1α, which are mitochondrial targets (Figures 3 A, B).
Figure 3
Immunohistochemical staining of HIF-1α and FASL. A – The upper panel in the figure demonstrates the expression of HIF-1α (pink) with reduced expression in the minocycline-treated group (M) compared to the vehicle (V) group (control). H represents the healthy group. The lower panel presents the expression of FASL, which shows that minocycline can reduce the FASL expression and regulate FAS-mediated cell death (400×). B – Quantification of HIF-1α and FASL positive cells
Values are expressed as mean ± SEM and were considered significant at p < 0.05 (*p < 0.05 vs. vehicle group, **p < 0.01 vs. vehicle group)
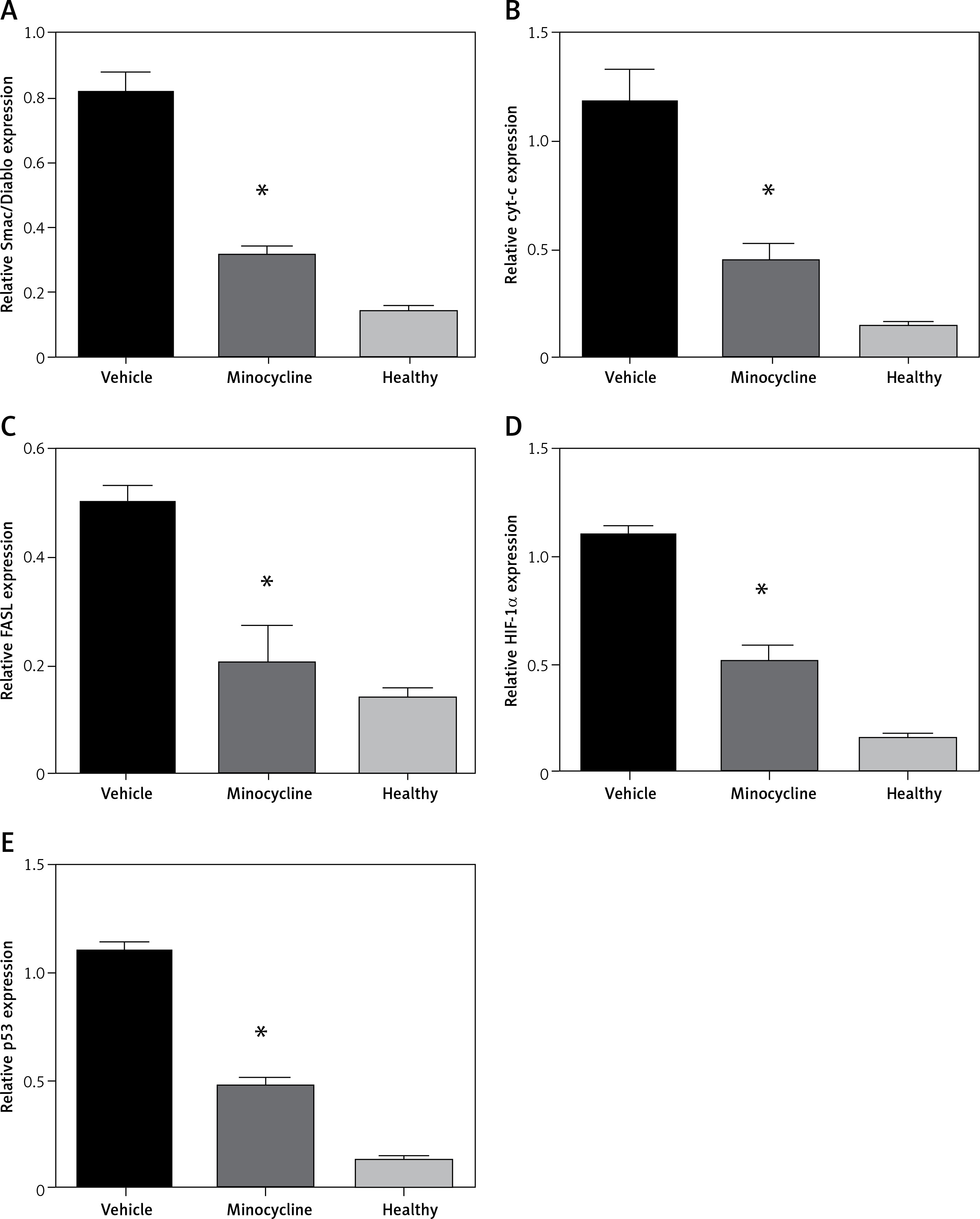
Evaluating mRNA expression of mitochondrial-dependent cell death genes and HIF-1α
To confirm the results obtained by ELISA, the mitochondrial-dependent genes (Smac/Diablo, cytochrome c), FASL and HIF-1α were subjected to qPCR. In addition, p53 gene expression was evaluated to confirm the involvement of HIF-1α in apoptosis. There was a consistent reduction in the expression of Smac/Diablo, cytochrome c, FASL, HIF-1α and p53 in minocycline-treated samples compared to the control (*p < 0.05 vs. vehicle, **p < 0.01 vs. vehicle) (Figures 4 A–E).
Figure 4
Relative quantification of mRNA. The graphs represent the relative mRNA expression of Smac/Diablo, cytochrome-c (cyt-c), FASL, HIF-1α, p53 (A–E). There was a significant reduction in expression in the minocycline-treated group compared to the vehicle group. All values are expressed as mean ± SEM and considered significant at p < 0.05
*P < 0.05 vs. vehicle group, **p < 0.01 vs. vehicle group
Quantifying the HIF-1α and FASL protein expression in mitochondrial-dependent cell death
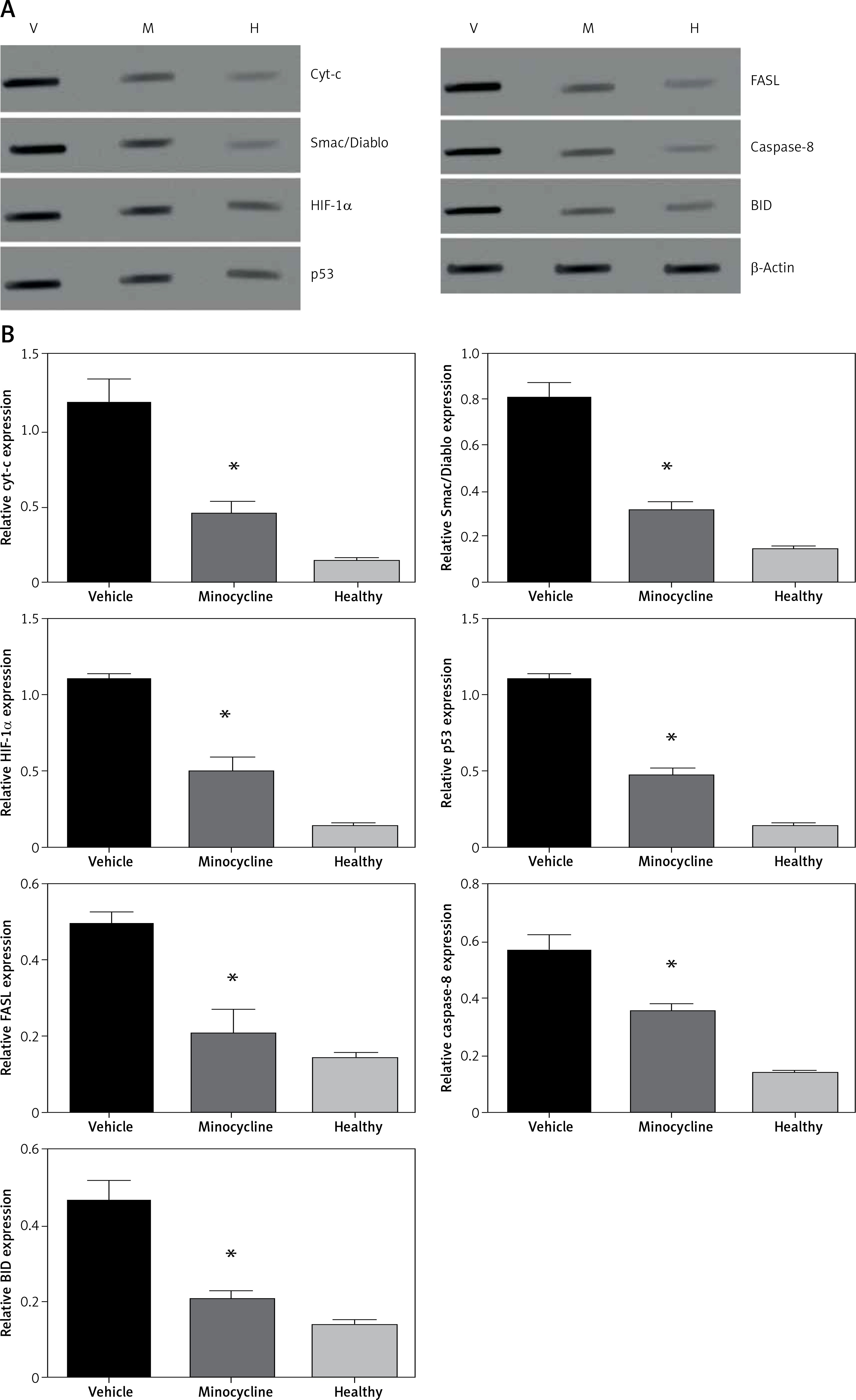
The treatment with minocycline consistently reduced the expression of mitochondrial-dependent genes (HIF-1α and FASL) associated with the cell death following spinal cord injury. The expression of HIF-1α and FASL was confirmed at the protein level and was investigated using western blot. To further corroborate the crosstalk between intrinsic and extrinsic pathways of apoptosis, expression of caspase-8 and BID was also determined. Caspase-8 cleavage and BID activation are required for the FAS-mediated apoptosis. Figures 5 A, B shows the western blot of proteins Smac/Diablo, cytochrome c, FASL, HIF-1α, p53 caspase-8 and BID.
Figure 5
Western blot analysis. A – The figure represents the western blot of mitochondrial targets where V represents the vehicle, M is the minocycline-treated group and H represents the healthy group. The results were consistent with the mRNA expression analysis of these targets. Caspase-8 and BID expression was included to evaluate the effect of the caspase-dependent pathway in the FAS-mediated cell death mechanism on treatment with minocycline. The images were visualised using Kodak Image Station software. B – Quantification of western blot analysis
All values are expressed as mean ± SEM and considered significant at p < 0.05. *p < 0.05 vs. vehicle group.
Discussion
The chief rationale behind this study was to investigate the effect of minocycline, a widely used anti-inflammatory drug, in inhibiting mitochondrial-dependent cell death mediated by FASL in association with HIF-1α. Various sources of evidence have revealed the efficacy of minocycline in alleviating neurological disorders including spinal cord injury [4, 6, 21]. Although the effect of minocycline in inhibiting cell death has been established previously, its effect on FASL-mediated cell death and HIF-1α following traumatic spinal cord injury is unclear. Mitochondria contain molecules that initiate the release of substances (cytochrome c and Smac/Diablo) leading to apoptosis that can either be caspase dependent or independent [7, 22]. The cytoprotective activity of minocycline is reported to be associated with mitochondrial transmembrane potential and blocking of calcium uptake [23].
Neuronal death through the FAS death receptor pathway is one of the principal events in spinal cord injury [13]. FASL mediates apoptosis through pro-inflammatory cytokines and previous studies have demonstrated that inhibition of FASL activity might improve the neurobehavioural recovery in animal models of brain ischaemia and spinal cord injury [24–26]. Spinal cord injury has also been reported to modulate the expression of HIF-1α. However, the effect of minocycline in regulating HIF-1α expression has not been studied so far. Reports suggest that the expression of HIF-1α following spinal cord injury is time dependent and has a chief role in promoting cell survival [16]. Our study brought together the effect of minocycline on FASL-mediated apoptosis and the expression of HIF-1α.
In the present study, the rats were administered with minocycline following spinal cord injury and its effect was evaluated compared to the vehicle. Cell death following traumatic spinal cord injury was evaluated through TUNEL assay and demonstrated that minocycline significantly reduced cell death compared to the vehicle group (Figure 1). Since our major objective was to evaluate the effect of minocycline on FAS-mediated apoptosis dependent on mitochondrial cell death, we performed an ELISA assay to measure the release of Smac (DIABLO), cytochrome c, HIF-1α, FASL and TNF-α. Smac/Diablo and cytochrome c function as death promoting factors released due to the rupture of inner mitochondrial membrane as a result of the pathophysiological cascade that follows spinal cord injury [27]. Minocycline has been shown to be efficient in inhibiting the release of these pro-apoptotic factors [6], which was confirmed in the current study too. The findings were also congruent with the previous literature where minocycline treatment potentially targets cyt-c to promote functional recovery following spinal cord injury [28]. Considering these actions together, i.e. mitochondrial activity, HIF-1α expression, and FASL-mediated apoptosis, we inferred that these activities were interlinked and studied the corresponding minocycline effect. We also quantified the release of TNF-α as FASL belongs to the TNF family and is linked to mitochondrial activity as well [29].
Minocycline reduced the FASL expression at the mRNA and protein level, which was confirmed by immunohistochemistry (Figure 3). HIF-1α expression was increased in the vehicle group and reduced approximately two-fold with the minocycline treatment. HIF-1α expression was extremely low in the normal group. The expression of HIF-1α in the central nervous system was first demonstrated in an ischaemic brain and was subsequently found to be differentially expressed in spinal cord injury. However, this effect is time dependent, and it was reported that there was a gradual decrease in HIF-1α expression after 3 days of spinal cord injury [30]. Transplantation of neural stem cells that express HIF-α was reported to promote repair following spinal cord injury [31]. Hence, the pro-survival or pro-apoptotic activity of HIF-1α is highly time-dependent, where HIF-1α expression in the later stages of injury exhibits post-conditioning effects to improve neuronal repair. Our study focused on the early therapeutic strategy following spinal cord injury using minocycline. HIF-1α expression was significantly reduced and the cell death mechanism involving HIF-1α is p53 dependent. It was reported previously that hypoxia leads to an increase in p53 and is partially dependent on HIF-1α upregulation [32]. Minocycline treatment reduced p53 and HIF-1α expression levels, reducing cell death, emphasizing the synergistic activity of p53 and HIF-1α in spinal cord injury induced cell death. In addition, cell death by activation of the FAS pathway is caspase as well as p53 dependent [33]. Hence, minocycline reduced cell death by targeting HIF and FAS pathways, possibly interlinked via caspase-dependent or -independent targets.
In conclusion, the present study, therefore, demonstrates the effect of minocycline on mitochondrial-dependent cell death and the expression of HIF-1α following spinal cord injury in rats. Minocycline reduced the FASL-mediated apoptosis, and a marked decrease in the release of factors associated with mitochondrial-dependent cell death was observed. Minocycline could target intrinsic and extrinsic apoptotic pathways in association with regulation of HIF-1α and FASL expression.