Introduction
Migraine belongs to the commonest disorders worldwide and it significantly lowers quality of life and work productivity, resulting in a high burden for patients, society and employers. Annual migraine-associated cost in the US is about USD 80 billion [1].
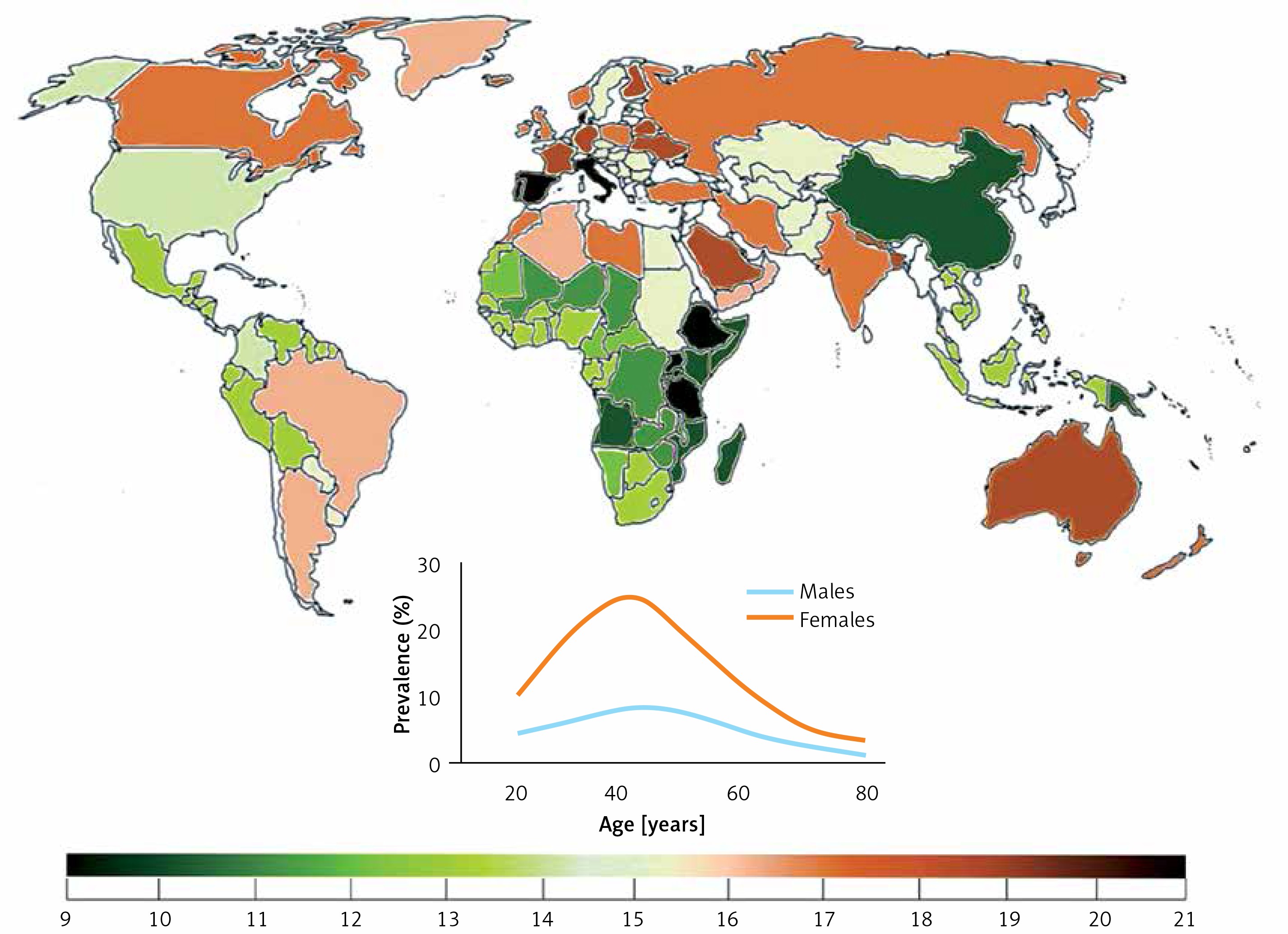
Migraine prevalence varies from continent to continent and from country to country, but in general it is relatively high as compared with many other chronic diseases. That prevalence depends on age and can be approximately three times higher in women than in men (Figure 1). Hover, the peak of migraine incidence is observed in adolescence. It is a complex disease affecting several regions of the brain, inducing vascular dysfunction, cranial vasodilation and release of some neuromodulators, such as dopamine [2]. Current migraine therapies are effective only in a proportion of patients and studies on new therapeutic targets are needed.
Figure 1
Worldwide migraine prevalence in 2016 according to WHO (both sexes, all ages). Inset – prevalence by age and sex (https://vizhub.healthdata.org/gbd-compare/)
Recurrent episodes of headache are the hallmark of migraine and they are usually associated with an array of other symptoms, including nausea, vomiting, photo- and phonophobia, visual disturbances and others. According to the International Headache Society, two main clinical subtypes of migraine can be considered: migraine with aura (MA) and without aura (MO). Other rarer type of migraine are basilar-type migraine (BTM) and familial hemiplegic migraine (FHM). Aura occurs in about one third of all migraine patients and includes mainly visual symptoms, but also aphasia, dyschromatopsia, prosopagnosia, ideational apraxia, alien hand syndrome and proper name anomia [3].
The pathophysiology of migraine is not fully known and there were two opposite theories on the cause and nature of migraine attacks – the vascular and the neuronal theory, which were then combined into the neurovascular theory [4]. Several other theories can be considered, including the hypothalamus theory, the serotonin theory, the depolarization theory and others. Migraine is a complex disease with the phenotype determined by the interaction between genetic, environmental and life-style factors. The pathogenesis of migraine is determined by phenomena occurring in the sensory cortex of the brain and/or the brain stem [5].
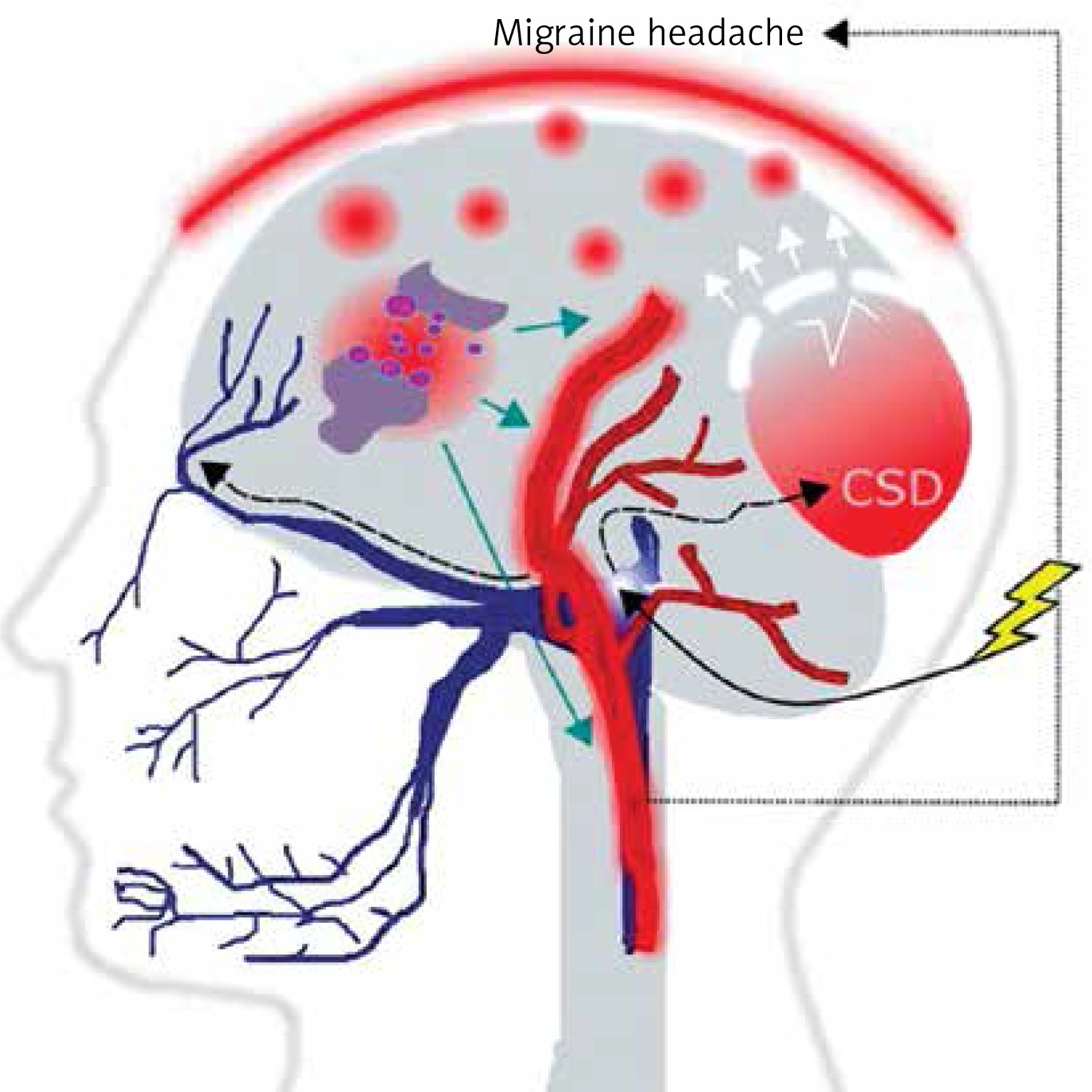
It is commonly accepted, although not firmly evidenced, that migraine is a threshold disease, released by a brain-related trigger [6–9]. Such migraine trigger and the wave of depolarization induce the activation of neurons of the trigeminal nerve, which release chemical substances inducing the dilation of blood vessels and inflammation, which directly cause migraine headache (Figure 2). Cortical spreading depression (CSD), a wave of neuronal hyperexcitability followed by suppression of neural activity, may be responsible for disturbances occurring in MA [10].
Figure 2
A migraine trigger (yellow thunder) acts on the nucleus (light blue oval) of the trigeminal nerve (dark blue) and activates it. Resulting waves of depolarization (black broken arrows) move along the nerve and reach the cortex, inducing cortical spreading depression (CSD). This results in an inflammation- like state and release of inflammatory neurotransmitters, which induce dilation of brain blood vessels (red), which in turn evokes migraine headache. This figure does not correspond to actual anatomical relationships within the brain
Early magnetic resonance imaging (MRI) studies revealed a low level of high energy phosphates in the brain of migraine patients, suggesting defects in brain energy metabolism in migraine [11]. These studies had several limitations. First of all they did not provide information on physiological dysfunctions associated with observed effects. Currently, more detailed imaging studies on migraine mechanisms are conducted with positron emission tomography (PET) and functional MRI (fMRI) (see [12] for review). Studies with these techniques showed that repeated migraine attacks modified metabolism in specific regions of the brain involved in sensory and pain processing, including the bilateral insula, premotor and secondary somatosensory cortex (Figure 3). These studies reported ATP depletion due to a deficient mitochondrial metabolism in brains of migraine patients [13]. These studies suggest a significant role of mitochondria in the pathophysiology of migraine.
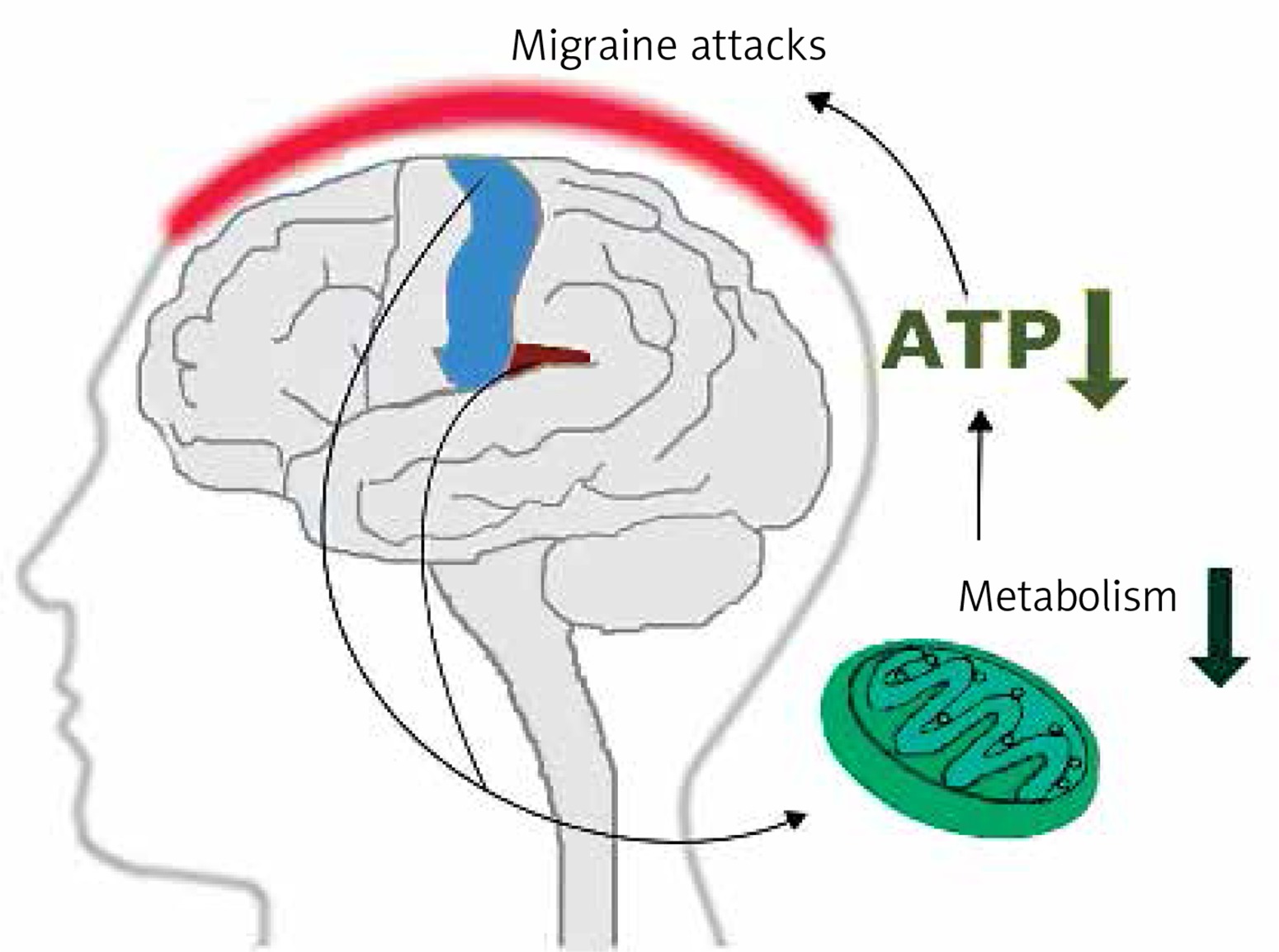
Mitochondria – an important player in migraine pathogenesis
Approximately three times higher frequency of migraine prevalence in females than males suggests either X-linked form of this disease, the mitochondrial transmission of another form of it, or both. However, if changes in mitochondria can contribute to the pathogenesis of migraine, not only mtDNA, but also the nuclear genes encoding mitochondrial components and regulating their functions may play a role.
Mitochondrial dysfunction can be associated with increased neuronal excitability resulting in increased susceptibility to migraine [14]. Any mitochondrial impairment in the brain likely leads to a deficit in mitochondrial energy, which in turn can be a migraine trigger. Some positive results on the influence of a high-fat and low-carbohydrate (ketogenic) diet on migraine and epilepsy course seem to support this assumption because such a diet stimulates mitochondrial metabolism [15, 16]. Further support is provided by the observation of the protective action of riboflavin and the Q10 coenzyme, both enhancing the function of the mitochondrial electron transport system (ETS, respiratory chain) against migraine [17–19].
A cross-sectional questionnaire-based study on 85 patients with various mitochondrial diseases (MDs) resulted in a higher frequency of headaches in this group compared to individuals without clinically detected mitochondrial impairments [20]. The highest prevalence of tension-type headache (TTH) was observed, but the prevalence of migraine and probable migraine was only slightly lower. This study was supported by research showing a higher prevalence of migraine in individuals with MDs than in the general population [21]. Stratification of migraine cases into MD types suggested that migraine might not be a phenotypic feature of a particular MD, but might instead be linked with a common susceptibility of the central nervous system to some factors, likely associated with defects in ETS accompanying that MD.
Riboflavin as well as MELAS and other mitochondrial diseases link migraine with mitochondria
Riboflavin (vitamin B2) belongs to the B complex vitamins playing an important role as coenzymes in many flavoprotein enzyme reactions. It occurs in two active forms, flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), which are important for vital mitochondrial processes, including metabolism of amino acids, purines and fatty acids as well as the redox reaction [22]. Riboflavin deficiency is a world-wide problem and it has been shown to improve the clinical picture of several mitochondrial diseases, including complex I and II-associated myopathies [23–29].
As brain metabolism is disturbed in migraine and riboflavin ameliorates the functioning of ETS, its evaluation in clinical trials as a prophylactic agent is justified [30]. Riboflavin prophylaxis was effective in migraine in adults, which may follow from the general neuroprotective effect of this compound [30–33]. Di Lorenzo et al. reported that migraine patients with non-H mtDNA haplogroup responded more frequently to riboflavin than patients with the H group [34]. On the other hand, the frequency of riboflavin non-responders was higher in the H group. There were no differences between MA and MO patients in riboflavin responsiveness. These studies suggest that the efficacy of riboflavin therapy can depend on the patient genotype.
Riboflavin decreased the frequency of headache attacks and their duration in a randomized, controlled trial performed by Shoenen et al. [35]. These results were confirmed in another clinical trial performed by Rahimdel et al., who also observed a decrease in frequency of headache attacks [36]. Several other clinical trials with riboflavin singly or in combination with other agents both in adult and adolescent migraine patients brought positive results – more details are presented in an excellent review by Thompson and Saluja [30].
Riboflavin was also reported to ameliorate mitochondrial dysfunction and protect against oxidative stress [22]. This compound protected against hyperglycemia and DNA damage in diabetic mice [37]. As we mentioned, the action of riboflavin may depend on the genotype of the recipient. Therefore, it is not very surprising that in some mitochondrial diseases which are associated with mutations, the efficacy of this drug can be low. However, riboflavin-based therapy is considered as effective or possibly effective at various levels of recommendation by several institutions, including the American Academy of Neurology and the American Headache Society (http://www.aan.com/guidelines).
Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) is a complex, progressive neurodegenerative disorder with a great variability in clinical picture causing difficulty with distinguishing it from vascular cerebral diseases, first of all cerebral infarctions [38]. Migraine shares some clinical characteristics with MELAS, including headaches with nausea and vomiting [21]. Another mitochondrial disease, myoclonic epilepsy with ragged-red fibers (MERRF), is also associated with episodes of hemicranial headaches. This provokes the question whether migraine can be a monosymptomatic form of MELAS or MERRF. However, no association of mutations typical for MELAS with migraine was reported. Klopstock et al. did not observe any association between MA and mutations (deletions and substitutions) common in MELAS and MERRF [39]. In a subsequent study, Buzzi et al. did not observe any association between the m.3243A>G substitution in the tRNALeu gene and pure matrilineal multigenerational MA or MO [40]. In addition, neither patient enrolled in that study carried this mutation. No association of the m.3243A>G mutation with MO was observed [41]. In another study no association between nine point mutations (substitutions) and migraine with prolonged aura was observed [42]. Therefore, variability of mtDNA typical for MELAS is not reported to occur in migraine, and so these results do not support the thesis on migraine as a monosymptomatic form of MELAS. However, these studies, although carefully designed, were performed on relatively small populations. Table I presents some diseases associated with migraine [43].
Table I
Some clinical conditions associated with migraine [43]
Energy deficiency is especially important in muscles and neural tissue, particularly when combined with other stress, and contributes to many symptoms of mitochondrial myopathies [44]. Such myopathies associated with neurological disturbances are called mitochondrial encephalomyopathies. Several neurological symptoms occur also in migraine, including headaches, hearing impairment and seizures [45]. Therefore, some mitochondrial encephalomyopathies and migraine can share similar pathways of pathogenesis and so it is reasonable to look for a genetic background of migraine in mtDNA [46]. Moreover, this association allows one to speculate on the connection between migraine and epigenetic changes, which are reported for migraine comorbid disorders [47, 48].
The human mitochondrial genome and its changes related to migraine
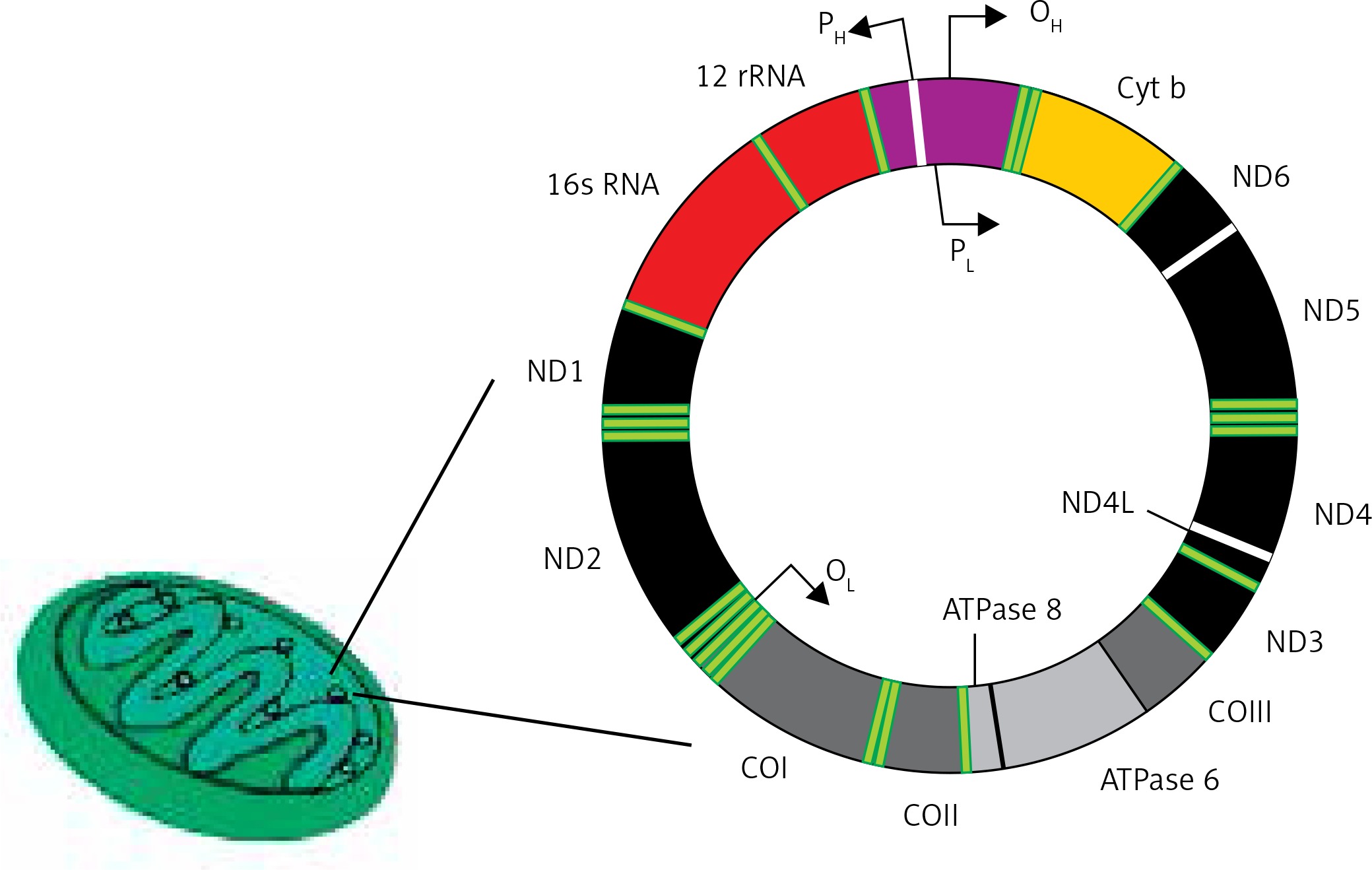
The human mitochondrial genome is a closed, double-stranded DNA (mtDNA) molecule of 16,569 bp, containing 37 genes encoding 2 rRNAs, 22 tRNAs and 13 polypeptides (Figure 4). All polypeptides encoded by mtDNA are subunits of ETS: 7 subunits of complex I (NADH: ubiquinone oxidoreductase), 1 subunit of complex III (ubiquinol:cytochrome c oxidoreductase), 3 subunits of complex IV (cytochrome c oxidase) and 2 subunits of complex V (ATP synthase). The entire complex III and all remaining subunits of complexes I, II, IV and V – 79 subunits in total – are encoded by nuclear DNA. Two strands of mtDNA are called heavy (H) and light (L) due to their base composition. mtDNA, in contrast to its nuclear counterpart, is densely packed with genetic information – more than 90 percent of it contains coding sequences, genes are intron-less and the ATPase6 and ATPase8 genes overlap. A small non-coding mtDNA fragment is called the control region, as it contains almost all control elements of mtDNA metabolism. Due to the high variability of human mtDNA its variants are classified into haplotypes [49].
Figure 4
Human mitochondrium is a double membrane-bound organelle found in the cytoplasm. It usually contains several copies of mtDNA, which is a closed double-stranded DNA having heavy (H) and light (L) strands. It has 13 polypeptide encoding genes – Cytb, ND1-6, ND4l, ATPase 6 and 8, COI-III – whose products are components of the mitochondrial electron transport chain. Other genes of mtDNA are 2 rRNA and 22 tRNA genes. Promoters (P) for these genes (2 for heavy strand and 1 for light strand) are located in a single regulatory region, where also the origin for heavy strand replication occurs (OH)
ETS is built from mitochondrial- and nuclear-encoded components and provides energy for the entire cell. However, even normally functioning ETS produces reactive oxygen species (ROS) and its malfunctioning increases ROS level. The importance and function of mitochondria should not be limited to energy production as this organelle plays an important role in calcium homeostasis and thermogenesis, the intrinsic pathway of apoptosis, signal transduction and other phenomena [50].
In humans, mtDNA does not recombine and is perpetuated exclusively in the maternal lineage. Therefore, it is useful for tracking genetic alterations through many generations. However, there are many mtDNA copies in a single cell, and not all of them need to be mutated to give a disease phenotype [51]. Therefore, mtDNA could also be exploited to track epigenetic changes sent from one generation to the next. However, epigenetic modifications to mtDNA have been far less exploited and are less well known than those to nuclear DNA.
mtDNA is characterized by greater sequence variation than its nuclear counterpart, but a cell usually has many mitochondria, which can have different genotypes. Therefore, wildtype mtDNA can occur along with its mutated variants in a single cell. This state is termed heteroplasmy in contrast to homoplasmy, which corresponds to the same sequence of a mtDNA locus in all mitochondria. Therefore, when a disease-associated variant occurs in a heteroplasmic form, its phenotypic consequences can be difficult to predict. In an extreme case, a lethal mutation in some mitochondria can be counterbalanced by the normal variant in remaining organelles. This creates a complex relationship between genetic variants in mtDNA and diseases. At a cellular level, a point mutation is considered to be expressed phenotypically with the threshold 80–90%, but for a larger deletion the threshold is lower – 50–60% [52]. At the tissue level, the proportion of mutants can positively correlate with the severity of the disease [53].
As mentioned, a strong connection between migraine and mitochondria was evidenced by the observation that migraine-affected individuals have impaired brain energy metabolism [54]. Mitochondria play a vital role in energy production, apoptosis regulation and production of ROS even in normal conditions [55]. These ROS are important in cell signaling, but their excess can lead to damage to cellular molecules [56]. The main source of ROS is oxidative phosphorylation (OXPHOS), producing ATP from ADP and inorganic phosphate (Pi), powered by the transfer of electrons from NADH/FADH2 to O2 [57]. This transfer is executed by mitochondrial ETS containing four large multiprotein complexes, designated I–IV, located in the inner mitochondrial membrane, each having several electron carriers. The fifth complex, ATP synthase, accepts protons transported from the inner mitochondrial membrane, which fuel ATP productions. Additionally, inner membrane-bound ubiquinone and cytochrome c, located in the intermembrane space, play an important role in OXPHOS.
Oxidative stress is considered as a factor in migraine pathogenesis and is associated with an excess of ROS, which can damage DNA. However, Geyik et al. found no difference in oxidative stress markers, total oxidant status (TAS) and oxidative stress index (OSI) between migraine patients and controls [58]. An increased level of 8-hydroxy-2’-deoxyguanosine (8-oxoG) in plasma of migraine patients was observed in that study and this increase was more pronounced in MO than MA patients. 8-oxoG is a product of oxidative DNA damage, which may lead to mutation if not repaired or misrepaired, and it is considered as an oxidative stress marker in humans. In similar studies, Alp et al. observed that serum TAS levels were lower in patients with MO as compared to controls and ROS levels in serum were higher in these patients [59]. Patients displayed higher values of OSI than controls, who, in turn, exhibited a higher levels of total SH groups, which were negatively correlated with headache duration. On the other hand, OSI was positively correlated with headache frequency. These studies have several limitations, including the small number of patients enrolled – less than 100 migraine cases in either study. Moreover, the studies were performed in a non-target tissue and the parameters for assessing oxidative stress were chosen rather arbitrarily and they do not decide about the occurrence and intensity of oxidative stress. Furthermore, a higher level of 8-oxoG can result from impairment in the DNA repair system or in general an impaired DNA damage response. 8-oxoG is mainly removed by the base excision repair (BER) system and in humans the hOGG1 glycosylase cuts the N-glycosylic bond between 8-oxoG and the DNA sugar [60, 61]. However, the results presented in those papers are a weak rationale to causatively correlate migraine with oxidative stress and allocate the source of this stress to mitochondria.
The brain is highly dependent on oxidative metabolism of glucose and therefore it is sensitive to changes in the functioning of mitochondria, which is supported by the association of several neurodegenerative diseases with mitochondrial dysfunction [62–64].
Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) is an important regulator of the expression of genes involved in mitochondrial biogenesis and cellular reaction to oxidative stress [65, 66]. Altered levels of mRNA of the PGC-1α gene along with a decreased number of mtDNA copies were observed in trigeminal neurons in a rat model of migraine [67].
Variability of mitochondrial DNA in migraine
The mitochondrial genome has limited autonomy as thousands of proteins which can be found in mitochondria are encoded in the nucleus. Therefore, the variability of nuclear genes involved in mitochondrial functioning may be important in migraine pathogenesis. The problem of genetic variability in migraine is connected with migraine hereditability. An identical twin study resulted in the conclusion of approximately 50% migraine hereditability [68].
A positive association between the m.4336A>G mutation, located on the tRNAGln , gene and sensorineural hearing loss and/or migraine was observed [69]. Wang et al. found an association between the occurrence of childhood cyclic vomiting, often considered as a migraine-like disease, and two rare heteroplasmic sequence variations containing 6 point and 2 length variants located in the mtDNA control region [70]. Two single nucleotide polymorphisms, m.16129G>A and m.16176C>T, in that region were associated with childhood cyclic vomiting. Moreover, these authors found that the homoplasmic variants located in the nt 16040-16188 segment of the highly variable region 1 (HV1) important in the regulation of mtDNA replication were associated with both cyclic vomiting syndrome and migraine without aura. In another study, Zaki et al. found an association between two mtDNA polymorphisms, m.16519C>T and m.3010G>A, and migraine headache and cyclic vomiting syndrome [71]. A higher, about three times, prevalence of migraine in individuals with the m.3243A>G mutation was reported [72]. This prevalence was observed in both MO and MA as well as in men and women separately. No association between the m.11084A>G polymorphism and migraine occurrence, whether MA or MO, in a Japanese population was found [73]. No difference between occurrence of the m.11084A>G mutation in MA and MO patients was observed in a Danish population [74]. This mutation was detected neither in migraine patients, MA or MO, nor controls. It has functional significance as it replaces adenine with guanine in the gene encoding the ND4 subunit of ETS complex I, resulting in the substitution of threonine with alanine. Interestingly, this mutation was reported to occur in a substantial fraction (about 25%) of Japanese migraineurs and was absent in tension-type headache patients and individuals with no migraine [75].
Mitochondrial haplogroup U, represented by the 12308A>G polymorphism, can play a role in migraine pathophysiology as this polymorphism was reported to associate with an increased risk of occipital stroke [76, 77]. Also other haplogroups in mtDNA can be directly or indirectly related to migraine. Marcuello et al. reported an association between the J haplogroup and efficacy of energy production by ETS [78]. As mentioned, migraine is associated with energy deficit, so that haplogroup and its polymorphisms of mtDNA may influence the susceptibility to migraine as well as to other diseases associated with energy deficit.
Generally, there are few studies on the association between mtDNA variants and migraine occurrence and these studies are usually performed on relatively small populations. Therefore, they should be continued on larger cohorts. Moreover, studies on variants of nuclear genes encoding mitochondrial components and influencing mitochondrial function should be further developed, especially as they are free from several difficulties in mtDNA research, including heteroplasmy requiring sequencing of the entire mitochondrial genome with a high coverage. Therefore, further studies on the role of mtDNA disturbances in migraine are justified and needed.
Epigenetics of mtDNA and its potential in migraine
Epigenetics deals with mechanisms of the regulation of gene expression, which are not directly associated with changes in DNA sequences. Three major epigenetic mechanisms are related to DNA methylation, histone modifications and RNA regulation.
Two aspects of epigenetic regulation in migraine pathophysiology can be considered. One is related to transient changes in epigenetic profile, which may result in changes in phenotype. The other is associated with such changes in the epigenetic pattern, which are perpetuated from one generation to the next and contribute to familial cases of the disease.
Epigenetic therapy, i.e. therapy targeting the epigenetic profile of specific genes, is considered in many human disorders, first of all cancer, in which several epigenetic modifiers are tested in phase I and II clinical trials [79]. Such therapy is also considered in migraine [80]. Valproate, a histone deacetylate inhibitor, has been successfully tested in prophylaxis of several classes of migraine [81–85]. However, its action cannot be unequivocally associated with an epigenetic mechanism as valproate is an anticonvulsive drug with several modes of action [86–88]. Moreover, it is not known how valproate acts on mitochondria.
Passaro et al. found a direct correlation between epigenetic markers of the H3 histone associated with genes involved in neuronal plasticity and CSD in rat brain [86]. This study suggests a potentially important role of epigenetic modifications in migraine pathogenesis, which was supported by subsequent studies [89–91]. Moreover, this importance has been recently confirmed by Gerring et al., who have identified 62 independent regions in DNA isolated from peripheral blood of migraine patients, which were specifically methylated [89].
Many migraine comorbid mitochondrial diseases are associated with changes in the epigenetic profile [46]. Therefore, it is justified to explore the role of epigenetic modifications to mtDNA in migraine pathogenesis. However, the issue of epigenetics in mitochondria is still a matter of debate, with some controversial aspects [92]. As mtDNA is not associated with histones, its epigenetic modifications are limited in comparison with its nuclear counterpart, but mtDNA occurs in many copies and each of them could have a different epigenetic profile. Therefore, DNA methylation and regulatory RNAs are major players in epigenetic control of gene expression in mitochondria. In humans, DNA methylation of the CpG islands located in the promoters of many genes is of a particular significance. It is induced by DNA methyltransferases (DNMTs) 1/2 and 3A/B [93, 94]. However, it should be stressed that mtDNA does not contain CpG islands, such as those targeted by DNA methyltransferases in the nucleus, but dispersed CpG dinucleotides occur in the mitochondrial genome [95]. Mitochondrial DNA methyltransferase 1 (mtDNMT1), an isoform of nuclear DNMT1, plays an important role in regulation of the expression of mitochondrial genes by modification of the mtDNA methylation profile [88]. This enzyme was reported to bind mtDNA in the region of the D-loop containing all three promoters needed for the transcription of mitochondrial genes, suggesting that mtDNMT1 can regulate gene expression. It was shown that DNMT1 interacted with CpG dinucleotides within mtDNA [96]. There is another DNA methyltransferase in the nucleus, DNMT3L, which, on binding with unmethylated lysines in H3 histone, guides DNMT3A/3B to DNA [97]. As there is no H3 histone in mitochondria, the potential of this enzyme seems to be lower than remaining DNA methyltransferases. However, DNMT3L is involved in DNA methylation in the nucleus, so it can play a role in the expression of genes whose products are important for mitochondrial homeostasis.
Another DNA methyltransferase, DNMT3A, was localized in the mitochondria of neurons and its decreased activity was reported to be associated with an abnormal mtDNA methylation pattern in amyotrophic lateral sclerosis, a progressive neurological disease [98]. However, it should be taken into account that in contrast to mtDNMT1, DNMT3A was not specifically targeted to mitochondria, but was found in these organelles. The identification of DNA methyltransferases operating in mitochondria shed light on the epigenetic mechanisms in these organelles, but on the other hand, it has added to the complexity of this issue [99]. Other epigenetics-related proteins detected in mitochondria are DNMT3B and two proteins involved in active demethylation, ten-eleven translocation (TET) 1 and 2 [100–103]. These methyltransferases are not only encoded in the nucleus, but are isoforms of nuclear proteins. Therefore, precise regulation of their expression and functions is required for cellular homeostasis and disease-related changes in their activity can result from an imbalanced nuclear-mitochondrial cross-talk [104]. However, some reports question the process of mtDNA cytosine methylation, indicating limitations of techniques employed in earlier studies [105].
The third aspect of epigenetics, RNA regulation, is first of all based on three kinds of non-coding (nc) regulatory RNAs: short ncRNA, including micro RNA (miRNA) of 22–23 bases in length (b) and piwi-interacting RNA (piRNA) of 26–31 b; medium ncRNA of 50–200 b and long non-coding RNA (lncRNA) longer than 200 b [106].
Micro RNA regulation is important in the nervous system [107, 108]. RNA interference components, including miRNAs and Argonaute proteins, were localized to mitochondria, showing another aspect of the cross-talk between the nucleus and mitochondria through an array of miRNAs, which are called “mitomiRs” [108].
Long non-coding RNAs are another element of epigenetic regulation, and in the nuclear genome it is associated with several important aspects of the regulation of gene expression, including chromosome X inactivation [109]. lncRNA regulation is important in many cancers, including brain tumors [110, 111].
The mitochondrial transcriptome is quite complex and should not be limited to 37 mtDNA genes, as almost the whole mitochondrial genome is transcribed – 99.9% of H and 97.6% of the L strand were reported to undergo transcription [112]. Many antisense transcripts can be found within the mitochondrial transcriptome. Moreover, many RNAs encoded in the nucleus are present in mitochondria and vice versa – several mitochondria-borne RNAs were reported to operate in the nucleus, indicating the importance of the mitochondria-nucleus cross-talk [113]. Altogether, many both “short” and “long” regulatory RNAs, including miRNAs and lncRNAs, can be found in mitochondria, and they can play an important role in the regulation of gene expression. In general, two species, miRNA and lncRNA, seem to be mutually regulated within the miRNA-lncRNA axis giving the functional balance between lncRNAs and miRNAs – the lncRNA-miRNA paradigm [114]. Departures from this paradigm can contribute to disturbances in energy production and can affect other processes in mitochondria important for the cell fate, which may result in disorders [115]. However, to our knowledge the role of lncRNA, either nuclear or mitochondrial, in migraine pathophysiology has not been explored so far. An association between disturbances in lncRNA functioning and mitochondrial impairment was observed in Down syndrome [116].
The mitochondria-nucleus cross-talk can also be expressed by the changes in the nuclear epigenetic profile reflected in mitochondria functioning, which in turn results in the global changes to the whole organism, as in the case of mitochondrial lifespan extension dependent on stress [117].
Several loci associated with migraine in the human genome have been identified in genome-wide association studies (GWAS) [118, 119]. Some of them can be linked with epigenetic mechanisms [120]. However, it should be underlined that no genes directly involved in establishing and maintaining the epigenetic profile, such as DNA methyltransferases or histone deacetylases, have been found in these studies. Moreover, GWAS studies did not provide results on the link between mtDNA variability and migraine.
Conclusions and perspectives
Epigenetic mechanisms are an important element of the control of gene expression. Transcriptional regulation through RNA molecules is essential for many genes [121, 122]. On the other hand, the epigenetic profile undergoes changes induced by environmental conditions. Oxidative stress is linked with pathophysiology of many diseases, including migraine. However, in most cases it is not completely known whether the stress belongs to the reasons or consequences of a disease, or both. It is difficult to find a disorder not linked to oxidative stress in its pathogenesis. On the other hand, ROS are normally produced in mitochondria. Therefore, many oxidative stress-related disorders can be potentially linked with mitochondria. Consequently, association of a mitochondrial dysfunction with a clinically detectable disorder may result from a false positive correlation. Oxidative stress, resulting from impaired functioning of ETS, may lead to the accumulation of damage to cellular components and mitochondrial aging, cellular senescence and premature organismal aging. So far, no mtDNA damage in migraine has been reported, but the extent of mtDNA damage in the brain of mammals was negatively correlated with maximal life span [22].
Abnormal expression of PGC-1α in neurons of a rat model of migraine provokes a question about the potential role of this protein in the pathogenesis of human migraine. Moreover, the involvement of PGC-1α in mitochondrial biogenesis prompts the search for the role of mitophagy, a key element in the regulation of mitochondrial number, in migraine pathogenesis [123].
Migraine can be diagnosed as an isolated headache with autonomic symptoms or as a part of a syndrome associated with other complex diseases, including mitochondrial disorders. There is no general agreement whether these two categories of migraine are different or not. Some reports suggest that there are essential differences between these two forms of migraine not resulting from individual susceptibility to this disease. These differences are claimed to manifest first of all with a prolonged aura and stroke-like episodes in migraine associated with other diseases. However, Vollono et al. recently observed migraine without aura in about 80% of mitochondrial disease cases [43]. Therefore, it may be difficult to distinguish autonomous migraine from migraine associated with other diseases.
Several mitochondrial encephalomyopathies share neurological symptoms with migraine, which may follow from a common mitochondrial element in their pathogeneses. As no migraine-specific mutations in mtDNA have been identified yet, it is likely that this common element can be of epigenetic nature. The brain needs energy in the form of ATP, produced mainly in mitochondria, and disturbance in its supply can result in an increased sensitivity to migraine attack.
Complex mitochondrial diseases cannot be related exclusively to mtDNA as they are associated with mitochondrial proteins encoded by nuclear genes. Therefore, a complex genomic analysis including both mitochondrial and nuclear components of the human genome should be performed in research on pathogenesis of these diseases. Mitochondrial variation need to be studied in large family pedigree studies to find a correlation between mtDNA mutation and migraine to eventually identify true causal variants. Moreover, these studies should also include variability in relevant nuclear genes. Currently, the MitoProteome Human Mitochondrial Protein Database lists 3,625 proteins, including isoforms and splice variants, which can be allocated to mitochondria (http://mitoproteome.org/).
Riboflavin proved to be effective in migraine prophylaxis in several clinical trials. However, some issues concerning the methodology of these trials as well as basic questions on riboflavin pharmacokinetics and pharmacogenomics should be addressed. These concern first of all interpretation of results of trials with n placebo groups and determination of optimal doses of riboflavin as the mechanism of its absorption is not completely known.
In clinical practice, it is important to distinguish migraine from monosymptomatic forms of other diseases manifested by headaches, first of all MELAS, especially considering that migraine can share morbidity with some mitochondrial disorders.
Mitochondria add complexity to migraine pathogenesis. Therefore, new integrated approaches are needed to better understand migraine pathophysiology and translate molecular research into clinical practice to improve diagnosis and therapy of this disease.
The question from the title of this paper remains open. First of all, we must extend and deepen our knowledge on epigenetic regulation in mitochondria. mtDNA has a specific structure, which can constitute a barrier in the determination of mtDNA epigenetic pattern with methods established for its nuclear counterpart, as was shown for cytosine methylation in the D-loop. Therefore, the initial enthusiasm associated with mtDNA methylation should be revised [118]. This is especially important as the functional relevance of mtDNA methylation has been supported only by association studies and no reliable mechanistic research on this subject has been performed. Secondly, although accumulating body of evidence suggests the involvement of mitochondria in migraine pathogenesis, the exact mechanism of this involvement is unknown. The potential role of mitochondrially enriched miRNAs (mitomiRNAs) in migraine pathogenesis should be investigated.
GWAS provided information on many loci which can be linked to migraine, so epigenome-wide association studies with several high-throughput analyses may bring more information on the role of epigenetics in migraine. However, the evaluation of the significance of mitochondrial genome/epigenome in migraine pathophysiology requires new methods and approaches.