

Coronavirus SARS-CoV-2
The SARS-CoV-2 virus, which is the responsible pathogen in the current pandemic, belongs to the Coronaviridae family. It is a single-stranded positive RNA virus with a long genome showing 80% similarity with SARS-CoV, and 50% similarity with MERS-CoV. The major part of the genome is associated with replicase-transcriptase encoding. Important proteins such as spike (S), envelope (E) and membrane (M) are expressed from subgenomic mRNA. The spike S protein is essential for infectivity of this particular virus type. Spike S protein is highly glycosylated and composed of two functional domains – S1 and S2. The viral entry to human cells is based on the reaction between the receptor binding domain (RBD) located on a spike protein subunit (S1) and the angiotensin converting enzyme II (ACE2) receptor present on human cells. There is an alternative way of human cells’ infection associated with expression of CD147 on the surface. CD147 is a transmembrane glycoprotein representing a co-receptor for different viruses, e.g. measles virus, HIV and SARS-CoV. The way is based on virus N protein bound to cyclophilin A interacting with the CD147 molecule, forming a complex. Moreover, it was shown that cyclophilin-CD147 interaction is involved in regulation of the inflammatory response. The lack of a sense of smell, a common symptom after infection with SARS-CoV, is a result of binding of the virus to the cellular receptor neuropilin-1 (NRP1) expressed on the olfactory epithelium. Different ways of infection with SARS-CoV-2 result in the variety of clinical symptoms and different course of disease [1, 2]. The SARS-CoV spike S protein is recognised as an antigen by APC (antigen presenting cells); in consequence of the immune humoral response, the specific antibodies are directed against this protein [2-4]. The localisation of the ACE2 receptor on different cells and tissues leads to development of a combination of clinical symptoms from the lungs, heart, gastrointestinal tract, kidneys, brain and others. The replication of this virus after the cell entry is based on translation from the genomic RNA by the replicase-transcriptase complex. This process requires ribosomal frameshifting. Moreover, in coronaviruses, the replicase-transcriptase complex is associated with remodelling of cellular membrane, facilitating further viral RNA synthesis as a result of this process [2].
Infection and clinical symptoms
Virus SARS-CoV-2 infection, like other viral infections, induces innate and adaptive immune responses with different effects on particular cells, tissue types and organs. The changes in numbers, phenotypes and functions of immunocompetent cells after SARS-CoV-2 infection are shown in Table 1. After binding of the ACE2 receptor by the SARS-CoV-2 virus, the activity and expression ACE2 on the cell surface decreases, leading to an imbalance of the renin-angiotensin system. It is the first step of changes in vessels, mainly in the lungs, resulting in increased permeability (leaky vessels), vasoconstriction, formation of platelet-fibrin microthrombi in small and medium size vessels (Fig. 1) with deep alveolar damage observed in patients with severe respiratory symptoms (acute respiratory distress syndrome – ARDS) [2, 5]. Disorders of the coagulation system and microthrombi formation in small and medium size vessels have been noted in many organs in relation to endothelial ACE2 expression. The study of inflammatory infiltrations in lung parenchyma of SARS-CoV-2 patients showed a mononuclear cell type of infiltration, containing mainly monocytes and lymphocytes. There were both T lymphocyte subpopulations (CD4+ and CD8+) with predominance of CD4+ cells and a high CD4 : CD8 ratio in consequence. CD4+ lymphocytes produce aggregates around changed small vessels, especially with microthrombi. CD68+ macrophages are present within the alveoli in increased numbers, forming infiltrations correlating with ARDS and disease severity. Lymphocytes and macrophages recruited to the lungs are activated, so they produce and release all types of cytokines, chemokines and other soluble mediators according to their potential and repertoire [5]. The clinical symptoms related to different organs after SARS-CoV-2 infection are shown in Table 2.
Fig. 1
Mechanisms of COVID-19 infection – vascularpathophysiology as an example (according to ref. [11])
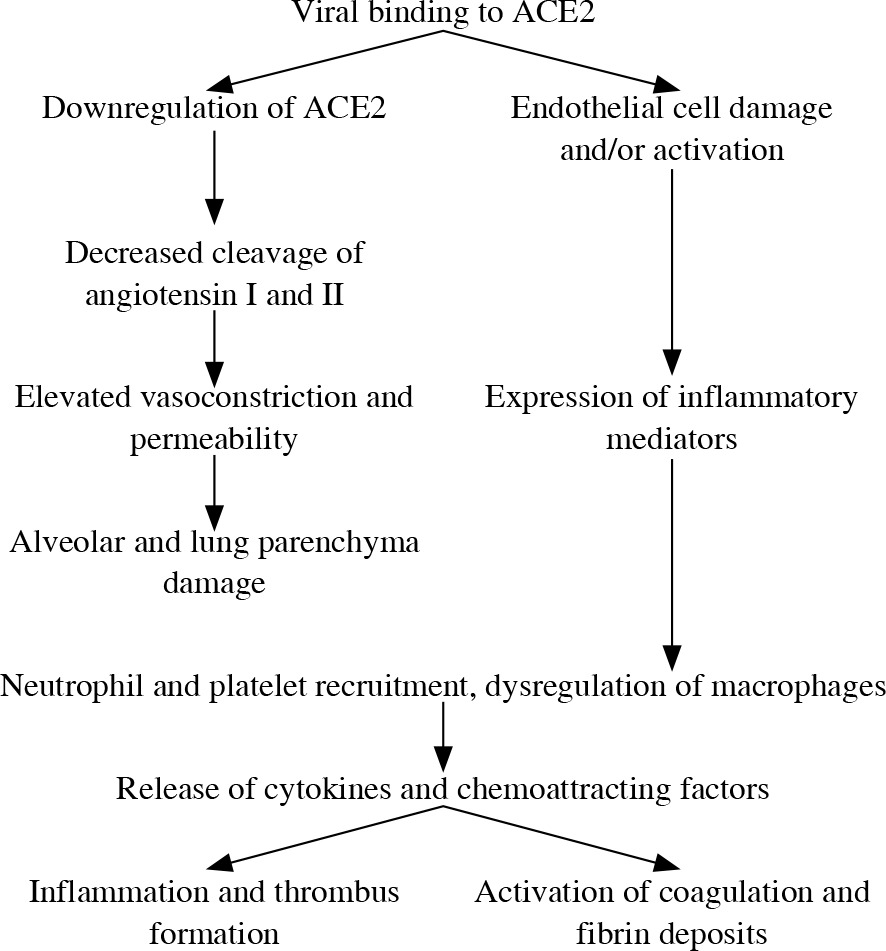
Table 1
SARS-CoV-2 infection – mechanisms and effects of infection on innate and adaptive immunity cells (according to ref. [2])
Table 2
Organ infection with SARS-CoV-2 – pathological findings leading to damage and impaired function (according to ref. [2])
Hyperinflammation and cytokine storm
The process of inflammation is important to clear infection of bacterial or viral aetiology. This inflammation is usually under control by the immune system. After elimination of the pathogen, the inhibition of cell activation, damping down the inflammation, returns the immune system to the state before stimulation. According to one hypothesis, called the “cytokine storm,”, uncontrolled overproduction of proinflammatory cytokines leads to a severe and critical course of disease with a fatal outcome in many cases, based on the cytopathic effect of SARS-CoV-2 infection. Infected cells die during viral replication. SARS-CoV-2 infection of epithelial cells causes the process of massive death of infected cells with a high level of pyroptosis, an inflammatory form of apoptosis (programmed cell death). It is suggested that pyroptosis may be an initiating factor for the hyperinflammatory response to infection (Table 3) [5]. Interleukin (IL)-1β is the main cytokine released during pyroptosis of infected cells. A high level of IL-1β was observed in serum and bronchoalveolar lavage fluid of patients with a severe course of infection. Moreover, epithelial cells infected with SARS-CoV-2 release viral RNA detected by other cells based on pattern recognition receptors (PRR). Activation of PRR and IL-1R stimulates and support secretion of proinflammatory cytokines and chemokines including IL-6, interferon γ (IFN-γ), MCP-1/CCL2, MIP-1a/CCL3, MIP-1b/CCL4, IP-10/CXCR4. Activated macrophages and lymphocytes (mainly T CD4+ cells) recruited to the place of infection sustain and potentiate an inflammatory response in tissue, e.g. lungs, resulting in damage of lung parenchyma [5]. The list of cytokines noted at high levels in serum of SARS-CoV-2 infected patients with a severe and critical course of disease includes IL-1β, IL-1RA, IL-7, IL-8, IL-9, IL-10, fibroblast growth factor (FGF), G-CSF, GM-CSF, IFN-γ, IP-10, MCP-1, MIP-1a, PDGF, and tumor necrosis factor (TNF) (Table 4). Additionally, in patients with severe disease, the level of VEGF was higher than in healthy people, stimulating neoangiogenesis and migration of activated immune cells and fibroblasts as an additional mechanism of alveolar injury [1, 2, 4, 5]. This profile of cytokines and chemokines was noted in many groups of patients, but in some cases, other cytokines were present at a high level. These observations showed IL-18, IL-33, TGF-β [3], IL-17, PD-L1, and IL-4 [6], but the clinical significance is not fully known. Hyperinflammation with hyperactivation and overproduction of cytokines are probably basic mechanism for exhaustion of T cells, NK cells and macrophages. However, the impaired function and exhaustion of immune system are not exclusive mechanisms of SARS-CoV-2 avoiding immune system control.
Table 3
Mechanisms of SARS-CoV-2 infection and evasion from immune system surveillance (according to ref. [1, 11])
Table 4
Clinical significance and prognostic role of cytokines and chemokines produced during SARS-CoV-2 infection (according to ref. [4])
Escape from immune system surveillance
The mechanisms of SARS-CoV-2 immune system evasion are not precisely described, but the similarity with SARS and MERS viruses suggestings some ways of escape. The common mechanism to avoid recognition is formation of a double vesicle shielding from PRR sensing. In SARS viruses, N protein from the surface is suspected to interfere with RNA recognition by the host. Another possible mechanism is typical for many pathogens – reduction of antigen presentation due to downregulation of MHC expression on APC [2]. The main antiviral cytokine, IFN-γ, directly inhibits viral replication and enhances antigen presentation. It is reported that SARS-CoV-2 synthesises and secretes a novel, short protein encoded ORF3b, inhibiting expression of IFN-β, which facilitates and enhances viral pathogenicity [3, 4]. Moreover, the synthesis of other protein – non-structural proteins (nsps) – was noted. Nsp16 methylates viral RNA, therefore preventing recognition and suppressing the type I IFN response [7].
NK cells
The NK cells are a small population (up to 10-15%) of circulating mononuclear cells with a crucial role in antivirus defence. Their main functions are production of cytokines and direct cytotoxicity of transformed cells with viral infection or the malignant process. NK cells develop in bone marrow and in secondary lymphoid structures such as tonsils, lymph nodes, and spleen. They belong to the group of innate lymphoid cells, preferentially type 1 (ILC). Steps of NK cell development and maturation in bone marrow and lymphoid tissues include expression of stage specific determinants (e.g. CD244 in precursors, CD122 in stage 2b of development, up to expression of CD16 and CD56 in mature NK cells and KIR in adaptive NK cells), and loss of determinants typical for early precursors (e.g. CD34). NK cells are often divided into circulating conventional NK cells (cNK) with ontogeny mainly in bone marrow and non-conventional NK cells localised in tissues (tissue resident NK cells – trNK). The differences between cNK and trNK are shown in Table 5. The main mechanisms of elimination of infected or transformed cells by stimulated NK cells is based on direct lysis of target cells. These NK cells are stimulated and directed against transformed cells due to upregulation of self-encoded molecules induced by infection, stress, downregulation of ligands for inhibitory receptors such as KIRs (killer cell immunoglobulin-like receptor), and direct recognition of viral moieties via PAMPS [7]. NK cell activity is modulated by cytokines, mainly stimulating – IL-2, IL-12, IL-15, IL-18 – alone or in combination, and IFN type I produced by infected cells or activated APC. These cytokines promote NK cell survival, proliferation, cytotoxicity and production of cytokines [7, 8].
Table 5
NK cells and viruses
NK cells are crucial in antiviral defence. There are two basic mechanisms of NK action – production of IFN and other stimulating cytokines inducing activation of T cells and direct cytotoxicity of infected cells [8, 9]. Despite the combination of cytokines, NK cells are activated by infected DC (dendritic cells) during cell-to-cell contact in draining lymph nodes. Stimulated NK cells produce IFN-g with double activity – promoting non-cytolytic control of virus replication and enhancing the antiviral T cell response. Production of IFN-g by activated NK cells is supported by IL-18 released by activated, inflammatory monocytes. Within many receptors associated with reaction with the virus-infected target, KIR2DL1/2 and NKG2A present on NK cells are important. Both receptors can be inhibited by interaction of viral proteins with HLA-C and HLA-E followed by binding to KIR2DL1/2 and NKG2A [8, 9]. The inhibitory receptor NKG2A interacts with the non-classical MHC receptor HLA-E, a potent regulator of NK cell antiviral activity. After recognition of an infected cell, NK cells become highly cytotoxic, leading to direct lysis of the target cell with perforin monomeric, granzyme and granulin. Moreover, NK are able to eliminate infected cells with another mechanism: CD16-mediated ADCC (antibody- dependent cell-mediated cytotoxicity). In this last way of infected cells’ elimination, antibodies to viral antigen and receptor FcgIII/II on the NK cell surface are involved. ADCC is possible only when target cells are antigen- opsonised with specific antibodies produced by B cells and NK cells are educated by self-specific HLA-binding receptors [8, 9]. The study of the antiviral response in mice showed a subpopulation of NK cells with memory properties. In humans memory NK cells were shown in a study of the response to specific viral antigens and cytokine profile. These memory NK cells express CD94/NKG2C, CD57, CG69, SYK, EAT2 and KIRD2DL markers [10].
NK cells and SARS-CoV-2
Circulating NK cells are recruited to SARS-CoV-2 infected tissues and organs, most frequently the lungs. These NK cells express high levels of CD16 and KIR, which means high cytotoxic activity and ability to lyse infected cells. In this point, the questions why activated NK cells are not able to eliminate the SARS-CoV-2 virus, and why the massive infection leads to a fatal outcome, are crucial. The answer is not simple. Observations in patients with SARS-CoV-2 infection showed an inverse correlation of circulating NK cell number and disease severity – in critically ill patients the NK cell number was lower than in patients with a mild course. Moreover, circulating NK cells in severe disease demonstrated a hyporesponsive phenotype with a low level of IFN-γ, TNF, IL-2 and granzyme production. On the other hand, it indicates that NK cells with a hyporesponsive phenotype do not contribute cytokines toward the cytokine storm. The NK cell number increased during recovery, suggesting transitional downregulation in the severe course. In lung tissue, in the mild course, the number of infiltrating NK cells was higher than in severe and critically ill patients. The decreased number of circulating NK cells during SARS-CoV-2 infection may be due to redistribution of these cells into infected tissues, despite NK cells’ exhaustion and death. There are other possible mechanisms resulting in hypoactivity of NK cells; one of them is associated with a high level of TGF-β noted in SARS-CoV-2 patients, and high levels of IL-6 and TNF in the hyperinflammatory state impairing cytotoxic and cytolytic function of NK cells. Another possible mechanism of inhibition of cytotoxic activity of NK cells is associated with modification of CD107a expression on NK cells after contact with lung epithelial cells transfected with S1 spike protein of SARS-CoV-2, which was observed in culture of NK cells with transfected Beas-2B cells. This co-culture resulted in decrease of CD107a expression (degranulation marker) on NK cells. The probable mechanism is based on expression of HLA-E on epithelial lung cells and interaction of viral proteins (including S1) with the NKG2A/CD94 inhibitory receptor on NK cells [11, 12]. Most of these observations came from the severe and critical course of SARS-CoV-2 infection. Studies of NK cell activity in patients with mild or asymptomatic SARS-CoV-2 infections are required, but they are difficult due to the low number of such patients [2, 5, 7, 13-15].
Summarising the study in SARS-CoV-2 patients, it seems that NK cells are ineffective due to virus-derived mechanisms leading to inhibition of cytotoxic and cytolytic activity. Considering the decreased number of NK cells in patients with a severe course of disease, the idea of using NK cells as a potential therapeutic target in SARS-CoV-2 infection was developed. So far, there are two clinical trials with NK cells used in SARS-CoV-2 infection: genetically modified NK cells (CAR-NK) in early-stage disease and CYNK-001 cells with NK cell activity (86 patients). The results are not yet published [14].
NK cells in lungs
The most interesting, in a SARS-CoV-2 infection, are resident lung NK cells and their activity against this particular virus. In physiology, NK cells account for up to 20% of lymphocytes within lung tissue. Considering the phenotype of lung NK cells, there are 3 subpopulations differing in combination of CD16 and CD103 expression. The function of lung resident NK cells during infection was shown in Mycobacterium tuberculosis infection with high release of IFN-γ and perforin with accumulation of these cells in lung tissue. During the infection, NK cells are recruited from blood into infected tissue but they still circulate in peripheral blood and further migrate after stimulation. The precise role of the lung resident NK subpopulation and percentage of circulating NK cells that become lung resident NK cells after migration are not known. Without infection the phenotype of NK cells is typical for mature CD56+, CD16-, CD57, NKG2D, KIR, CD49a cells without activation markers. Alveolar macrophages are probably responsible for the hypofunctional state of lung resident NK cells according to mechanisms regulating homeostasis [7, 12, 16-20].
NK cells in pneumonia
Experimental research with viral infections of lungs (respiratory syncytial virus, parainfluenza virus, influenza A virus) showed that NK activities blocked the virus burden and protected against severe and fatal disease. However, the non-active state of NK cells in lungs without infection suggested that prolonged highly active NK cells during lungs’ viral infection may potentiate lung injury, worsening clinical symptoms. The basic molecule exacerbating lung inflammation is IFN-γ at a high level [15, 20]. In Mycoplasma pneumoniae interstitial pneumonia the total number of NK cells and percentage of cytotoxic active NK cells CD158b+ which bind to MHC-I class determinants on target cells are lower than in control subjects. The number of NK cells recovers after a long time, e.g. 40 days, of the disease [15]. Observations of the influenza pandemic in 2009 showed infections of DC, macrophages and NK cells through surface sialic acid leading to inhibition of activation and minimal production of IFN-g, TNF and other pro-inflammatory cytokines. Moreover, influenza viral proteins are recognised by NCR NKp46 of NK cells, leading to direct lysis of target cells and stimulation of cytokine production. Evading immune control, influenza viruses developed the ability to change glycosylation of these proteins, limiting recognition by immune cells. Like other viruses, the influenza virus is able to redistribute MHC I proteins, enhancing interaction with inhibitory receptors on NK cells [21]. The in vitro study of the NK cell response to the influenza virus showed specific memory of these cells, suggesting significant anti- viral activity of the lung resident NK cell population in a new contact with the influenza virus [20].
SARS-CoV-2 infection of alveolar epithelial cells with massive virus replication and severe alveolar damage recruits immune cells to the site of infection. In lungs the NK cell population consists of two compartments – resident lung NK cells and NK cells coming from peripheral blood. The study of NK cells showed the adaptive NK phenotype mainly, with high expression of CD57+ and CD16+. The subpopulation of CD56bright NK cells was decreased in number. Expression of NKG2D, DNAM-1 and NKp30 was reduced. Bronchoalveolar lavage showed a lower NK cell number in the severe course of SARS-CoV-2 infection, and a higher number in the mild course. The number of NK cells in severe infection was similar to the number in healthy people. In critically ill patients, the number of NK cells in peripheral blood was deeply decreased, which, in consequence, diminishedreduced the possibility of migration of NK cells from blood into lungs [22, 23]. However, despite the low number of NK cells, expression of perforin on NK cells was significantly increased in the severe course of infection. It was important for the suggestion of a compensatory increase of cytotoxicity [22]. Hyperactivity of NK cells with overproduction of IFN-γ is associated with supporting and increasing lung inflammation. On the other hand, a prolonged hyperactivation state of NK cells makes them prone to exhaustion and cytolysis after killing infected target cells as a possible explanation for reduction of NK cell number [12]. In phenotype assay NK cells in patients with severe SARS-CoV-2 pneumonia and ARDS showed low expression of CD107a, increased expression of NKG2A, PD-1, CD39 and exhaustion markers – LAG3, PDCD1, HAVCR2 [7]. The lack of NK cells’ effective anti-viral function is an important immunological signal for macrophage activation syndrome. As back regulation in macrophage activation syndrome (hemophagocytic lymphohistiocytosis – HLH) high levels of IL-6 and IL-10 cytokines inhibit cytotoxic activity, perforin and granzyme expression of NK cells, finally eliminating NK cells as an important player in viral infection [12]. A study of patients who recovered from SARS-CoV-2 infection showed a slow increase of NK cells with no significant changes in expression of perforin and granzyme [22].
The immune systems (innate and adaptive) defence against viral infection is complex with a variety cells involved in different stages and aspects of the immune response. Despite pathogen elimination as the main goal of immune system activation, the long lasting cell memory (T, B lymphocytes and NK cells) as a potential fast and effective immune response against recurrent infection is a problem. In SARS-CoV-2 infection, the memory T, B and NK cell subpopulations and specific antibodies were observed, but the duration of cellular memory is unknown.