Introduction
Diabetes-associated vascular complications are associated with increased morbidity and mortality worldwide [1]. Notably, diabetic cardiomyopathy (DCM), which is characterized by left ventricular systolic and/or diastolic dysfunction, is a major diabetic complication. Several mechanisms responsible for the progression of DCM have been proposed, including hyperglycemia-mediated myocardial cytotoxicity [2–5], cardiomyocytes apoptosis [2–5], oxidative stress [2–5], and inflammatory reactions [4, 5]. These pathogenic developments commonly regulate the activation of mitogen-activated protein kinase (MAPK) [2], leptin [3], and nuclear factor-κB (NF-κB) [4] pathways. However, the inhibition of these signaling pathways does not prevent DCM, indicating that other mechanisms might contribute to the development of DCM.
Leptin, secreted by adipocytes and cardiomyocytes [3], has been implicated in the regulation of appetite, feeding-related behaviors, and energy metabolism. Leptin predominantly decreases food intake and increases energy consumption. Acumulating evidence has consistently demonstrated that leptin is involved in the development of atherosclerosis [6–8], systemic inflammation [7, 9], cardiovascular dysfunction [10], cardiomyocyte hypertrophy [11], and cardiomyocyte injury [3]. Leptin exerts effects via leptin receptor (Ob-R) and downstream signaling cascades, including extracellular signal-regulated kinase 1/2 (ERK1/2) [12], c-Jun N-terminal kinase/stress-activated protein kinase (JNK) [13], p38 MAPK [3, 14], proprotein convertase subtilisin/kexin type 9 [15], and Janus kinase 2 (JNK)/signal transducer and activator of transcription 3 (STAT3) [16]. Furthermore, in diabetes, leptin binds to the Ob-R to promote diabetes-related cardiovascular complications via p38 MAPK [3] and melanocortin-4 receptor signaling pathway [17]. Previous studies have reported that the JAK2/STAT3 pathway is also involved in hyperglycemia-mediated cardiovascular complications [18–20]. However, the effects and associated mechanisms of the JAK2/STAT3 pathway on hyperglycemia-induced cardiomyocyte injury and inflammation remain unknown. Furthermore, whether leptin plays a key role in hyperglycemia-induced cardiomyocyte injury and inflammation and is dependent on the JAK2/STAT3 pathway is unclear.
Naringin (4’,5,7-trihydroxyflavonone-7-rhamnoglucoside, NRG) exerts a variety of biological effects, including antioxidative, anti-apoptotic anti-inflammatory, and anti-diabetic effects [2–5]. In addition, NRG has cardioprotective properties. Our previous studies have shown that NRG protects cardiomyocytes from hyperglycemia-induced injury by suppressing the MAPK pathway, leptin and NF-κB. A recent study has also demonstrated that NRG suppresses hyperglycemia-mediated cardiac fibrosis via downregulation of protein kinase C (PKC) and p38 signaling pathways [21]. Similarly, we also found that NRG protects cardiomyocytes by upregulating KATP channels in vitro and inhibiting the NF-κB pathway in vivo and in vitro. The protective effects of NRG against ankylosing spondylitis (AS) are through suppression of the JAK2/STAT3 pathway in mice [22]. In contrast, NRG improves cognitive function in MHE/DA-treated animal models via activation of the JAK2/STAT3 pathway [23]. Therefore, we hypothesized that NRG might protect cardiomyocytes (H9c2 cardiac cells) against high glucose (HG)-induced injury and inflammation by regulating the JAK2/STAT3 pathway.
Material and methods
Material and agents
NRG, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1), dichlorofluorescein diacetate (DCFH-DH) and Hoechst 33258 were obtained from Sigma-Aldrich (St Louis, MO). Leptin antagonist (LA), leptin receptor antagonist (ObRa), and AG490 were purchased from ProSpec (East Brunswick, NJ). A Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies (Kumamoto, Japan). Fetal bovine serum (FBS) and Dulbecco’s modified Eagle’s medium (DMEM) were purchased from Gibco BRL (Grand Island, NY). Anti-STAT3, anti-phospho-STAT3, anti-JAK2, anti-phospho-JAK2, anti-leptin, and anti-leptin receptor antibodies were supplied by Cell Signaling Technology (Boston, MA), and horseradish peroxidase (HRP)-conjugated secondary antibody and bicinchoninic acid (BCA) protein assay kits were obtained from the KangChen Bio-tech, Inc. (Shanghai, China). Enhanced chemiluminescence (ECL) solution was purchased from the KeyGen Biotech (Nanjing, China).
Cell culture and treatments
H9c2 cardiac cells were provided by the Sun Yat-sen University Experimental Animal Center (Guangzhou, China). Cells were cultured in DMEM supplemented with 10% FBS under an atmosphere of 5% CO2 at 37ºC, then preconditioned with 80 μM NRG for 2 h before exposure to HG for 24 h. To further determine whether the protective effects of NRG and activation of the leptin-JAK2/STAT3 pathway were mediated by HG, cells were preconditioned with 50 ng/ml LA for 24 h or 25 ng/ml ObRa for 2 h or 10 μM AG490 for 30 min prior to exposure to 35 mM glucose for 24 h, respectively. After the treatments, cells were obtained for the following measurements and evaluations. Measurements of apoptosis, intracellular reactive oxygen species (ROS) generation, and matrix metalloproteinase (MMP) dissipation were performed at a cell density of approximately 1 × 106/ml.
Western blot analysis
Following those treatments, cardiac cells were obtained for western blot analysis. Protein expression was tested as follows. First, cardiac cells were lysed, and proteins were quantified using the BCA kit. Next, loading buffer was added to cytosolic extracts and boiled for 5–10 min, and the same amounts of cytosolic extracts were fractionated and transferred into polyvinylidene difluoride (PVDF) membranes. The PVDF membranes were blocked with fat-free milk, incubated with related antibodies (anti-total [t]-STAT3, detecting all forms of STAT3, 1 : 1000 in dilution, anti-phosphorylated [p]-STAT3, 1 : 1000 in dilution, anti-total [t]-JAK2 : detecting all forms of JAK2, 1 : 1000 in dilution, anti-p-JAK2, 1 : 1000 in dilution, anti-leptin, 1 : 1500 in dilution, anti-leptin receptor, 1 : 1500 in dilution, or anti-GAPDH, 1 : 2000 in dilution) in freshly prepared Tris-buffered saline with Tween-20, and then incubated with secondary antibody (HRP-conjugated goat anti-rabbit antibody). Finally, the immunoreactive signals were visualized by ECL detection. In order to quantify the expression level of each protein, the X-ray films were scanned and calculated using ImageJ 1.47i software (National Institutes of Health, Bethesda, MD). The protein levels of p-STAT3 and p-JAK2 were normalized to t-STAT3 and t-JAK2, respectively; the expression levels of leptin and leptin receptor were normalized to GAPDH. Experiments were carried out in triplicate.
Measurement of cell viability
Cardiac cells were obtained for the evaluation of viability as follows. First, cardiac cells were incubated with CCK-8 solution for 1.5 h. Next, absorbance at 450 nm was analyzed using a microplate reader (Molecular Devices, Sunnyvale, CA), and the mean optical density (OD) of each well in the indicated groups was used to calculate the percentage of cell viability based on the following formula: cell viability (%) = (ODtreatment group/ODcontrol group) × 100. The experiments were repeated five times.
Measurement of apoptosis
Apoptosis in cardiac cells was measured by Hoechst 33258 staining. The measurement was performed as follows. First, cells were washed with PBS, dried, and fixed with 4% paraformaldehyde. Then, the cells were seeded with Hoechst 33258 (5 mg/ml) at 37 °C, washed again with PBS, and dried. Finally, the cells were observed under a fluorescence microscope (BX50-FLA; Olympus, Tokyo, Japan). The experiments were repeated five times.
Measurement of intracellular ROS generation
Intracellular ROS generation was measured as follows. First, cells were washed with PBS, seeded with DCFH-DA solution (10 μM), and cultured for 30 min. Treated cells were washed again with PBS and dried, and finally, were observed using a fluorescence microscope connected to an imaging system (BX50-FLA, Olympus, Tokyo, Japan). The mean fluorescence intensity (MFI) was tested via Image J 1.47i software. The experiment was repeated five times.
Measurement of MMP
MMP was measured as follows: first, the treated cells were washed with PBS; second, cells were seeded with JC-1 (1 mg/l); third, the cells were washed again with PBS and dried; finally, the cells were observed under a fluorescence microscope connected to an imaging system (BX50-FLA, Olympus). The MFI of JC-1 was analyzed using ImageJ 1.47i software. The experiments were repeated five times.
Statistical analysis
All data are shown as the mean ± standard error of the mean (SEM). Differences between groups were analyzed by one-way analysis of variance using SPSS 16.0 software (SPSS Inc., Chicago, IL), followed by the least-significant difference post hoc comparison test. Statistical significance was considered for two-sided p-values less than 0.05.
Results
HG enhanced leptin and ObR expression in cardiac cells
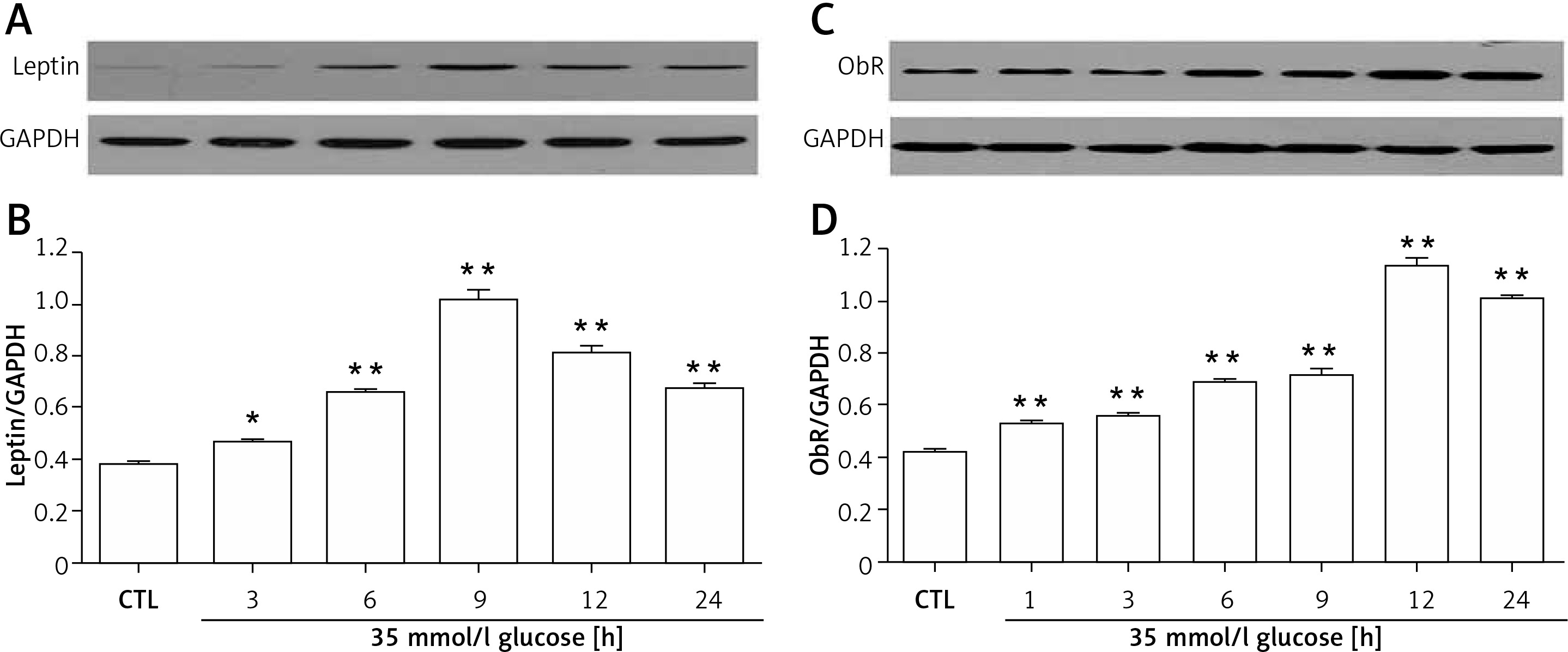
H9c2 cells were treated with HG for different times (3, 6, 9, 12, and 24 h), and as shown in Figure 1, leptin expression began to significantly increase after exposure to HG for 3 h, which reached the maximal level at 9 h (Figures 1 A and B). Similarly, the expression of ObR gradually increased over time, peaking at 12 h (Figures 1 C and D).
Figure 1
Expression of leptin and ObR in HG-induced H9c2 cardiac cells. A, B – Leptin expression began to significantly increase after exposure to HG for 6 h, and reached the maximal level at 9 h. C, D – ObR was gradually elevated over time, peaking at 12 h
Data are shown as the mean ± SEM (n = 3). **P < 0.01, compared with the control group.
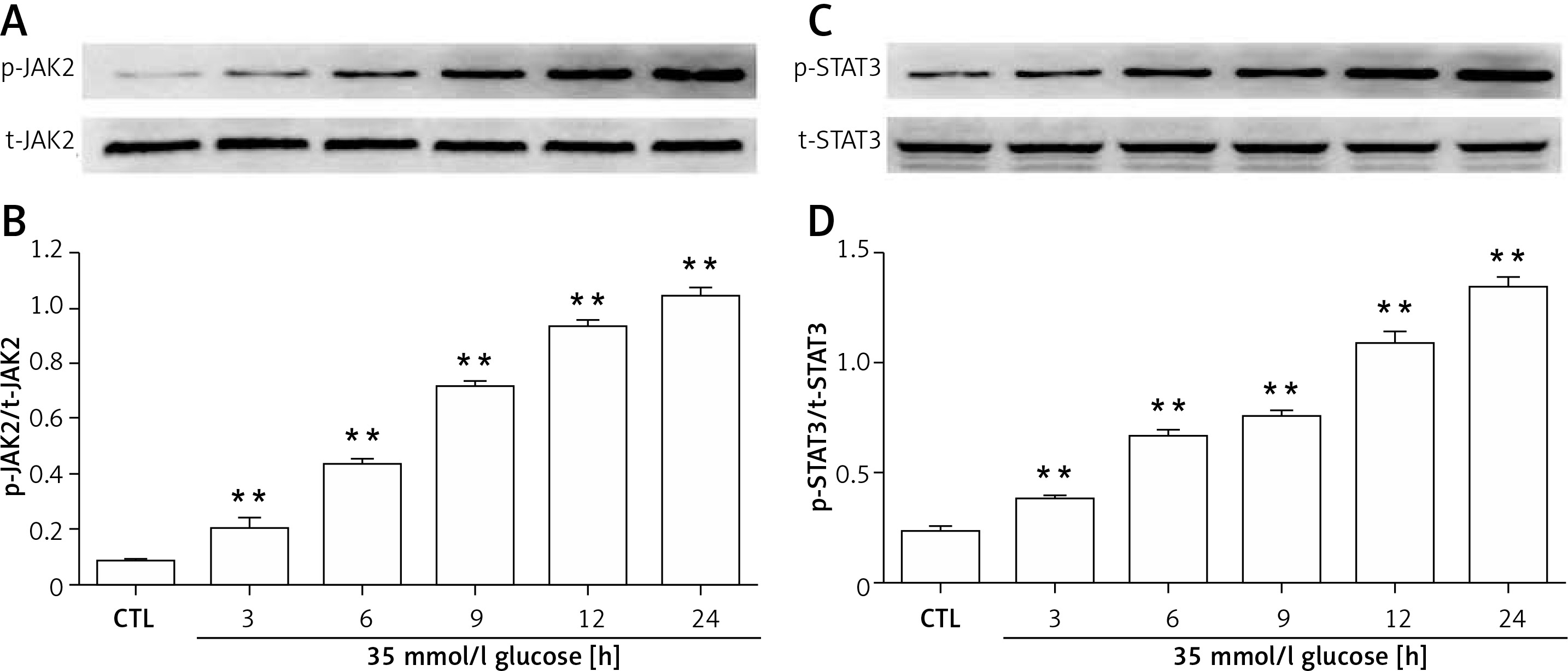
HG enhanced p-JAK2/STAT3 expression in cardiac cells
To evaluate the effect of HG on the JAK2/STAT3 pathway in cardiac cells, a time-response study was performed. As shown in Figures 2 A and B, the expression of p-JAK2 began to significantly increase after exposure to HG for 3 h, reaching the maximal level at 24 h. Similarly, p-STAT3 expression gradually elevated and peaked at 24 h (Figures 2 C and D).
Figure 2
Expression levels of both p-JAK2 and p-STAT3 in HG-induced H9c2 cardiac cells. A, B – p-JAK2 began to significantly elevate after exposure to HG for 3 h, reaching the maximal level at 24 h. C, D – p-STAT3 gradually increased, peaking at 24 h
Data are shown as the mean ± SEM (n = 3). **P < 0.01, compared with the control group.
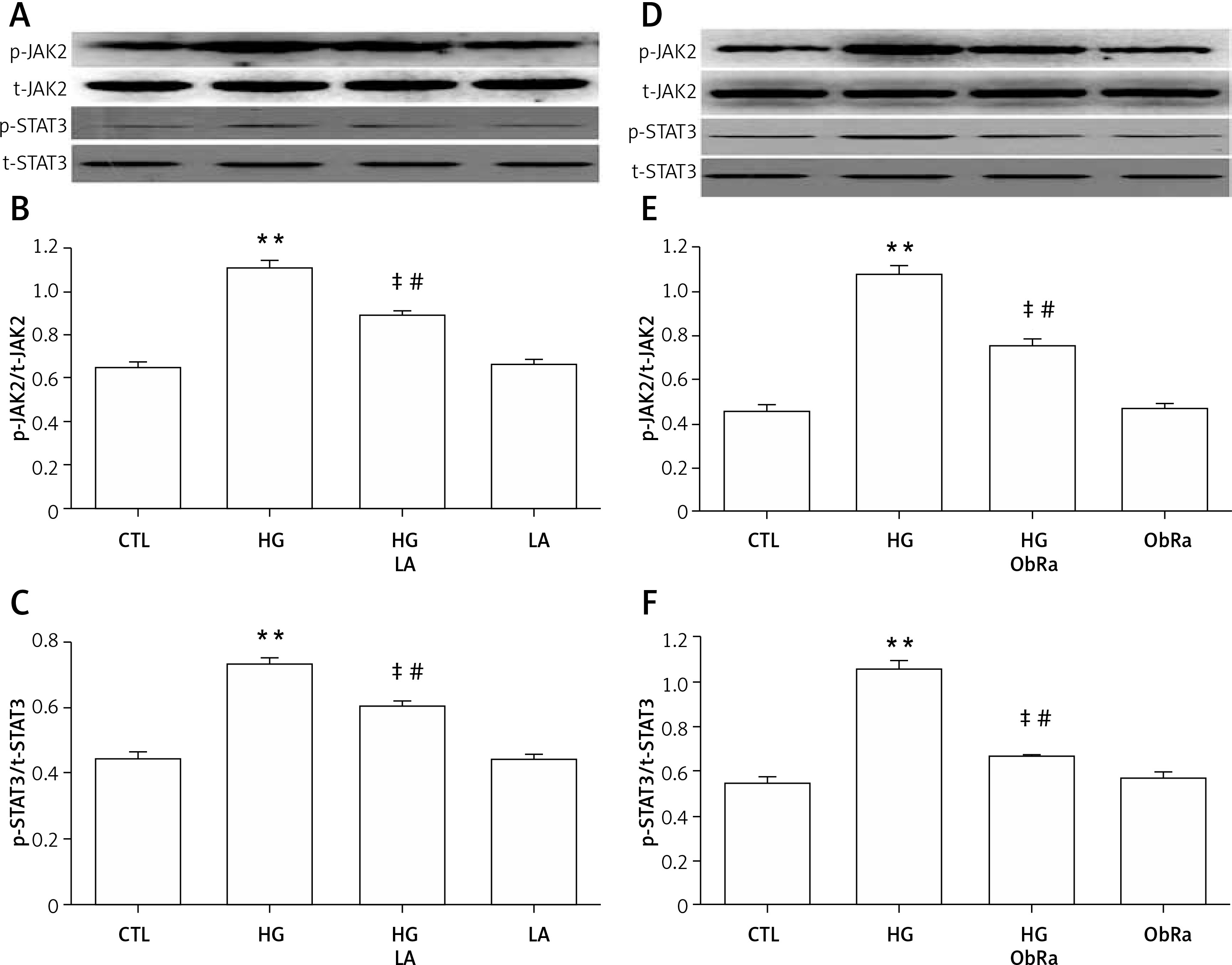
HG-induced p-JAK2/p-STAT3 upregulation was blocked by an LA or ObRa
Cardiac cells exposed to HG increased p-JAK2 and p-STAT3 expression levels (Figures 3 A–F). However, upregulation of p-JAK2 and p-STAT3 was markedly blocked by treatment with an LA (Figures 3 A–C) or ObRa for 2 h (Figures 3 D–F) prior to HG exposure.
Figure 3
Effect of LA and ObRa in H9c2 cardiac cells. H9c2 cells were divided into four groups according to treatment (control group, HG group, HG + LA/ObRa group, LA/ObRa group). Cells were treated with 35 mmol/l of glucose for 12 h in the absence or presence of pretreatment with either 50 ng/ml of LA for 24 h or 25 ng/ml of ObRa for 2 h. Levels of p-JAK2/t-JAK2 and p-STAT3/t-STAT3 were determined by western blot analysis. A–C – Upregulation of p-JAK2 and p-STAT3 in HG-induced H9c2 cardiac cells was markedly blocked by an LA prior to exposure to HG. D–F – Upregulation of p-JAK2 and p-STAT3 in HG-induced H9c2 cardiac cells was markedly blocked by ObRa prior to exposure to HG
Data are shown as the mean ± SEM (n = 3). **P < 0.01, compared with the control group. ‡P < 0.01, compared with the HG-treated group. #P < 0.05, compared with the control group.
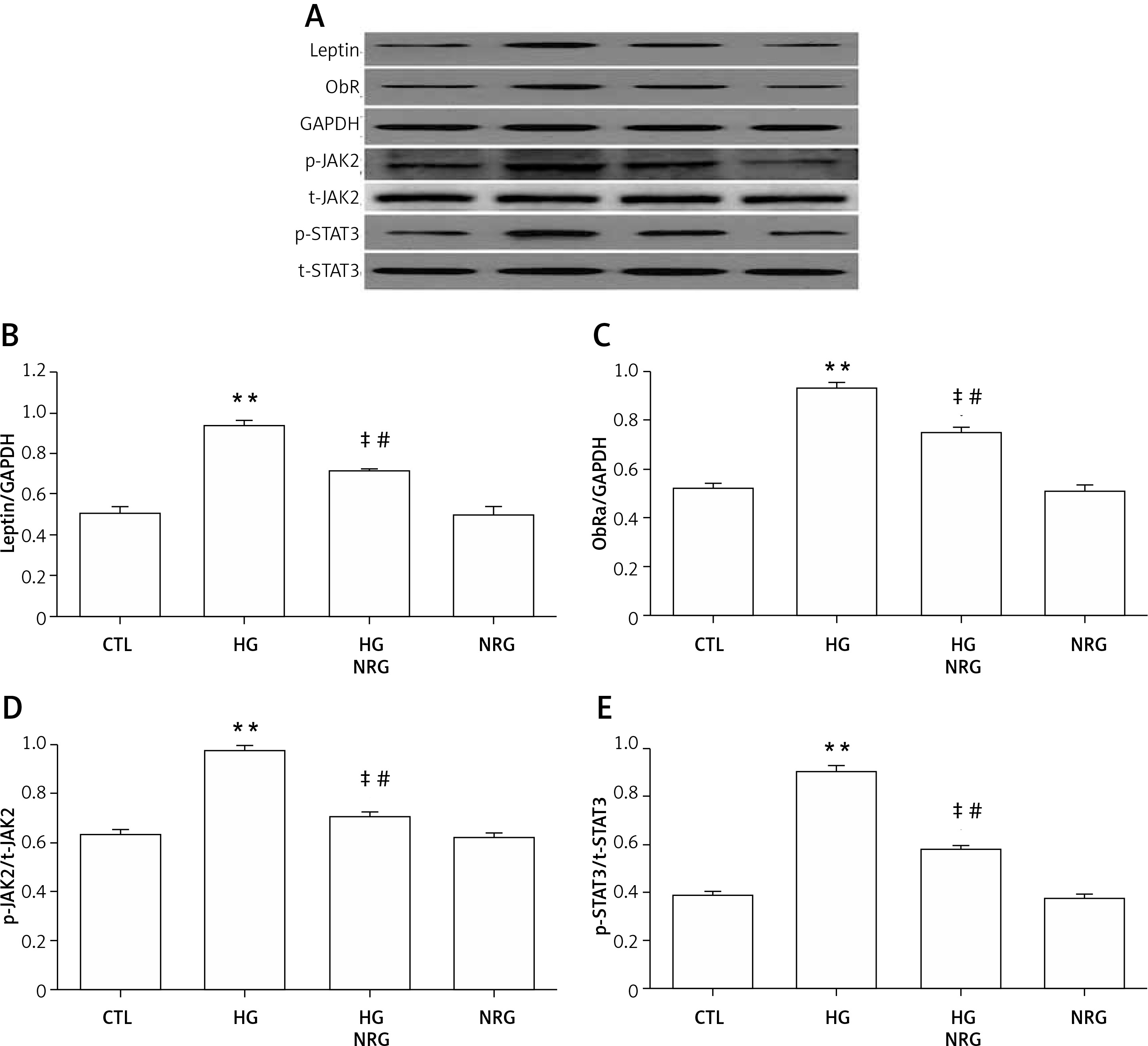
NRG ameliorated the HG-induced upregulation of leptin, ObR, and p-JAK2/p-STAT3 in cardiac cells
H9c2 cells exposed to HG markedly enhanced the expression of leptin (Figures 4 A and B), ObR (Figures 4 A and C), p-JAK2 (Figures 4 A and D), and p-STAT3 (Figures 4 A and E). However, the increased expression of each of these was significantly reduced by 80 μM NRG.
Figure 4
Effect of NRG in H9c2 cardiac cells. H9c2 cells were divided into four groups (control group, HG group, HG+NRG group, NRG group). Cells were treated with 35 mmol/l glucose for 9 h (for testing leptin), 12 h (for testing ObR), or 24 h (for testing p-JAK2 and p-STAT3) in the absence or presence of pretreatment with 80 μmol/l of NRG for 2 h prior to exposure to HG. Levels of leptin, ObRa, p-JAK2, and p-STAT3 were determined by western blot analysis. A–E – Levels of leptin, ObRa, p-JAK2, and p-STAT3 in the HG group were higher than those of the control group, and the increased expression was significantly reduced in the HG + NRG group compared with the HG group, which indicated that NRG ameliorated the HG-induced upregulation of leptin, ObRa, and p-JAK2/p-STAT3 in cardiac cells
Data are shown as the mean ± SEM (n = 3). **P < 0.01, compared with the control group. ‡P < 0.01, compared with the HG-treated group. #P < 0.05, compared with the control group.
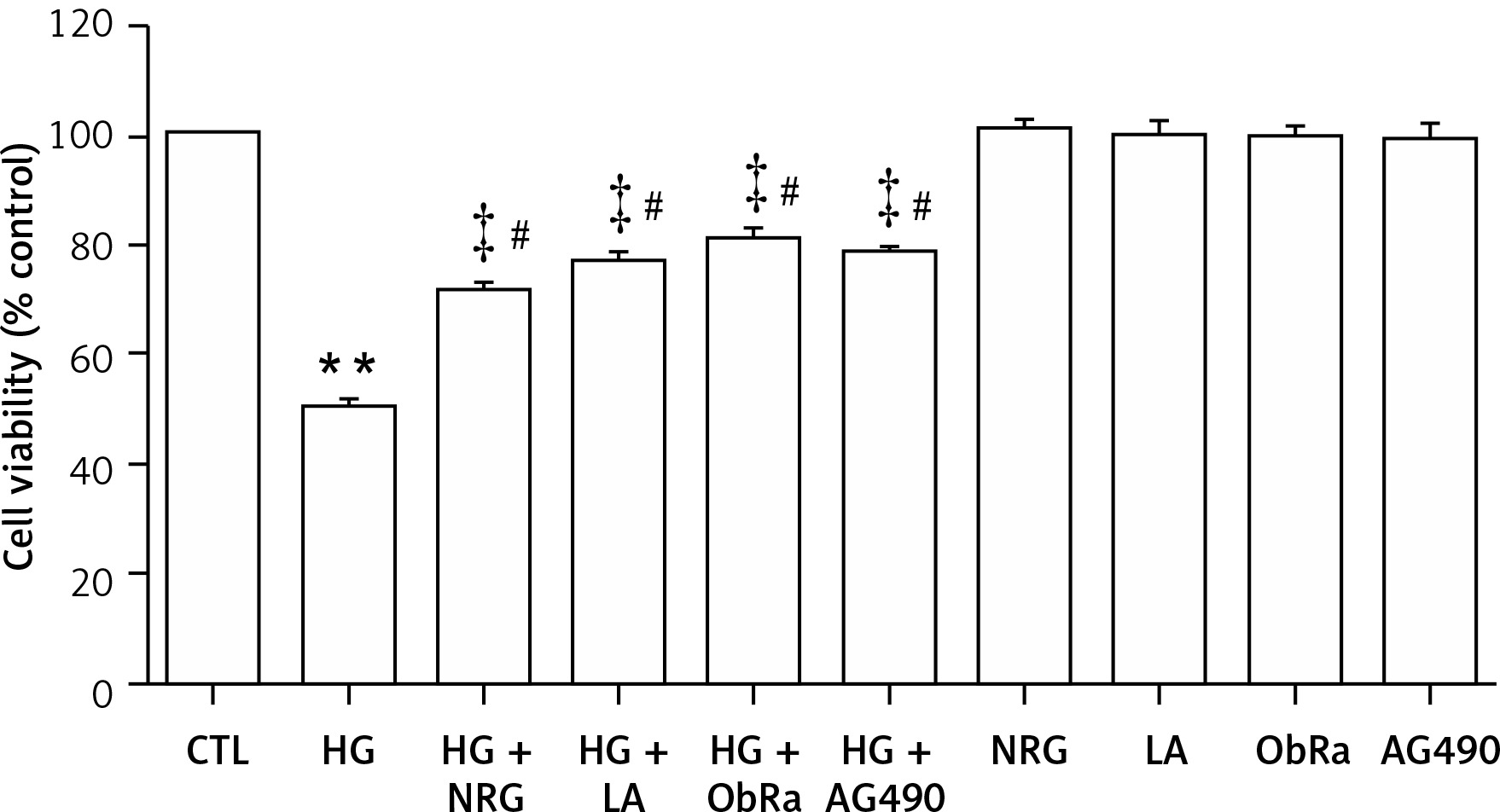
NRG and leptin-JAK2/STAT3 pathway inhibitors suppressed cytotoxicity in HG-treated cardiac cells
As shown in Figure 5, treatment of H9c2 cells with HG led to a significant decrease in cell viability. The adverse effects were inhibited by pretreatment with 80 μM NRG. Similarly, pretreatment of the cells with LA for 24 h, ObRa for 2 h, or 10 μM AG490 (an inhibitor of JAK2/STAT3) for 30 min also inhibited HG-induced cytotoxicity.
Figure 5
NRG, LA, ObRa, and AG490 attenuated cytotoxicity in HG-induced H9c2 cardiac cells. H9c2 cells were divided into 10 groups (control group, HG group, HG + NRG group, HG + LA group, HG + ObRa group, HG + AG490 group, NRG group, LA group, ObRa group, AG490 group). Cells were exposed to 35 mmol/l of glucose for 24 h in the absence or presence of pretreatment with 80 μmol/l of NRG for 2 h, 50 ng/ml of LA for 24 h, 25 ng/ml of ObRa for 2 h, or 10 μmol/l of AG490 (an inhibitor of STAT3) for 30 min prior to exposure to HG. Cell viability was detected by CCK-8 assay. HG-induced cell cytotoxicity and a significant decrease in cell viability was suppressed by pretreatment with NRG, LA, ObRa, or AG490
Results are presented as the mean ± SEM (n = 5). **P < 0.01, compared with the control group. ‡P < 0.01, compared with the HG-treated group. #P < 0.05, compared with the control group. LA – leptin antagonist, ObRa – leptin receptor antagonist, AG490 – an inhibitor of JAK2/STAT3 pathway.
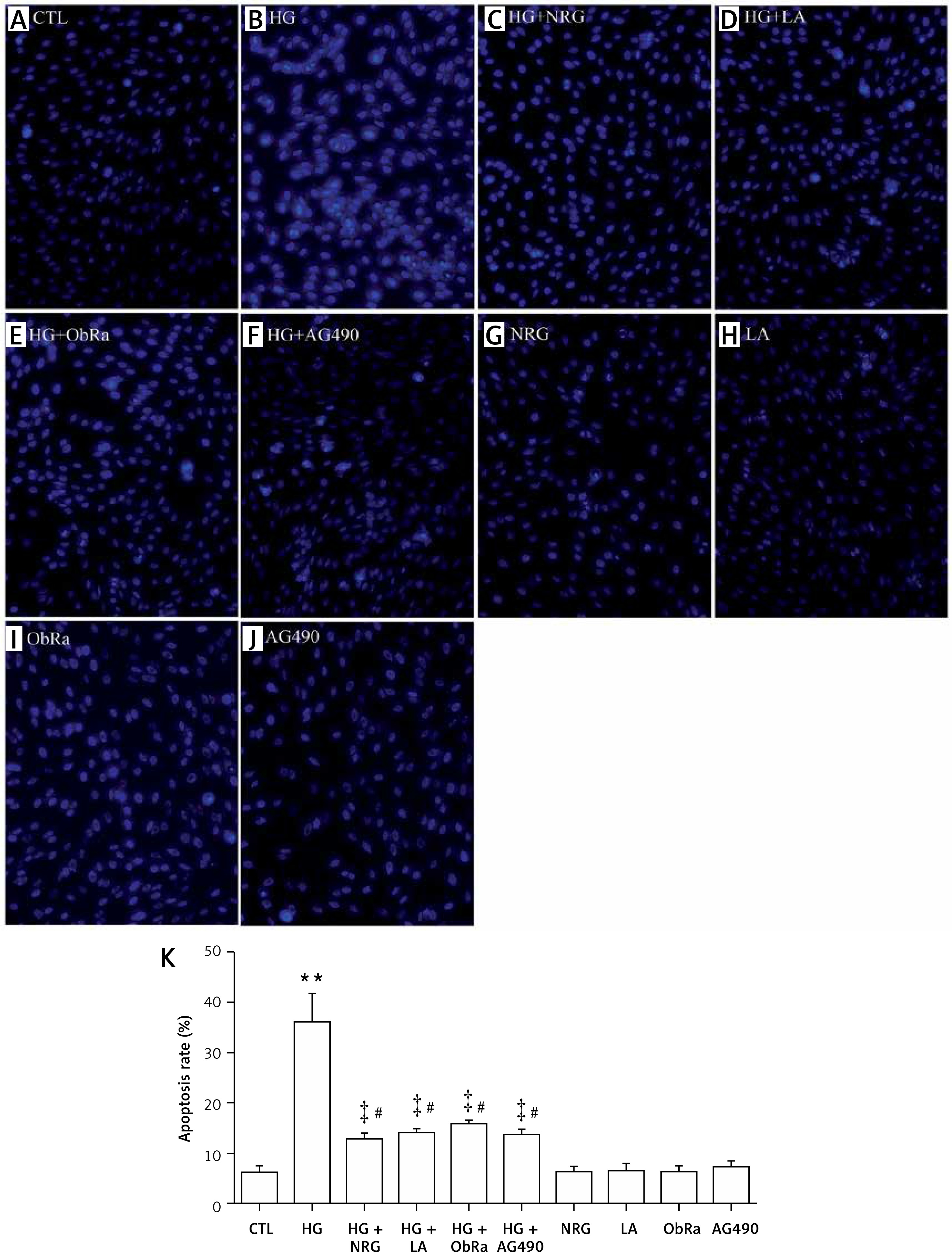
NRG, LA, ObRa, and AG490 ameliorated apoptosis in HG-treated cardiac cells
Results of Hoechst 33258 nuclear staining showed that HG caused apoptosis (Figure 6 B) when compared to the control group (Figure 6 A). Pretreatment of the cells with 80 μM NRG for 2 h prior to HG exposure considerably alleviated HG-induced cell apoptosis (Figures 6 C and K). Similarly, pretreatment of the cells with LA for 24 h (Figures 6 D and K), ObRa for 2 h (Figures 6 E and K), or AG490 for 30 min (Figures 6 F and K) also markedly reduced apoptosis (Figure 6 K).
Figure 6
NRG, LA, ObRa, and AG490 diminished apoptosis in HG-induced H9c2 cardiac cells. A–J – Hoechst 33258 nuclear staining followed by photofluorography to observe cell apoptosis. A – Control group (CTL). B – H9c2 cardiac cells were exposed to 35 mmol/l of glucose for 24 h. C – Cells were pretreated with 80 μmol/l of NRG for 2 h prior to exposure to HG for 24 h. D – Cells were pretreated with 50 ng/ml of LA for 24 h prior to exposure to HG. E – Cells were pretreated with 25 ng/ml of ObRa for 2 h prior to exposure to HG. F – Cells were pretreated with 10 μmol/l of AG490 for 30 min prior to exposure to HG. G – Cells were treated with 80 μmol/l of NRG for 2 h. H – Cells were treated with 50 ng/ml of LA for 24 h. I – Cells were pretreated with 25 ng/ml of ObRa for 2 h. J – Cells were pretreated with 10 μmol/l of AG490 for 30 min. K – Quantitative analysis of MFI using ImageJ 1.47i software (A–J)
Data are shown as the mean ± SEM (n = 5). **P < 0.01, compared with the control group. ‡P < 0.01, compared with the HG-treated group. #P < 0.05, compared with the control group. NRG – naringin, LA – leptin antagonist, ObRa – leptin receptor antagonist, AG490 – an inhibitor of JAK2/STAT3 pathway.
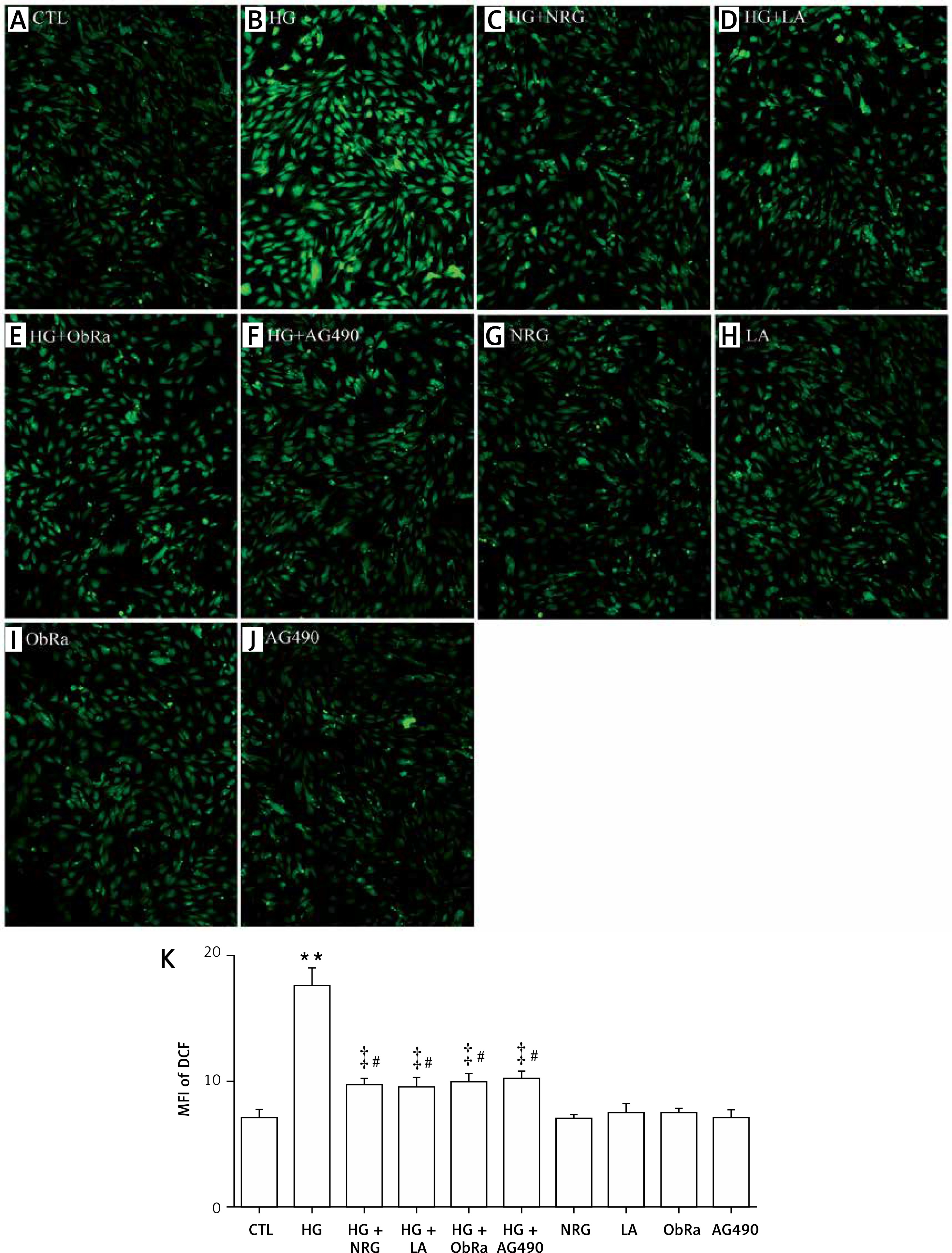
NRG, LA, ObRa, and AG490 suppressed oxidative stress in HG-induced cardiac cells
As shown in Figure 7 B and K, the exposure of cardiac cells to HG for 24 h increased intracellular ROS levels when compared to the control group (Figure 7 A). Pretreatment with 80 μM NRG for 2 h prior to exposure to HG markedly mitigated HG-triggered ROS production (Figures 7 C and K). Similarly, pretreatment with LA for 24 h (Figures 7 D and K), ObRa for 2 h (Figures 7 E and K), or AG490 for 30 min (Figures 7 F and K) also diminished the accumulation of ROS.
Figure 7
NRG, LA, ObRa, and AG490 decreased ROS production in HG-treated H9c2 cardiac cells. A–J – DCFH-DA staining followed by photofluorography to observe the intracellular ROS level. A – Control group. B – Cells were exposed to 35 mmol/l of glucose for 24 h. C – Cells were pretreated with 80 μmol/l of NRG for 2 h prior to exposure to HG for 24 h. D – Cells were pretreated with 50 ng/ml of LA for 24 h prior to exposure to HG. E – Cells were pretreated with 25 ng/ml of ObRa for 2 h prior to exposure to HG. F – Cells were pretreated with 10 μmol/l of AG490 for 30 min prior to exposure to HG. G – Cells were treated with 80 μmol/l of NRG for 2 h. H – Cells were treated with 50 ng/ml of LA for 24 h. I – Cells were pretreated with 25 ng/ml of ObRa for 2 h. J – Cells were treated with 10 μmol/l of AG490 for 30 min. K – Quantitative analysis of MFI using ImageJ 1.47i software and the data in A–J
Data are shown as the mean ± SEM (n = 5). **P < 0.01, compared with the control group. ‡P < 0.01, compared with the HG-treated group. #P < 0.05, compared with the control group. NRG – naringin, LA – leptin antagonist, ObRa – leptin receptor antagonist, AG490 – an inhibitor of the JAK2/STAT3 pathway.
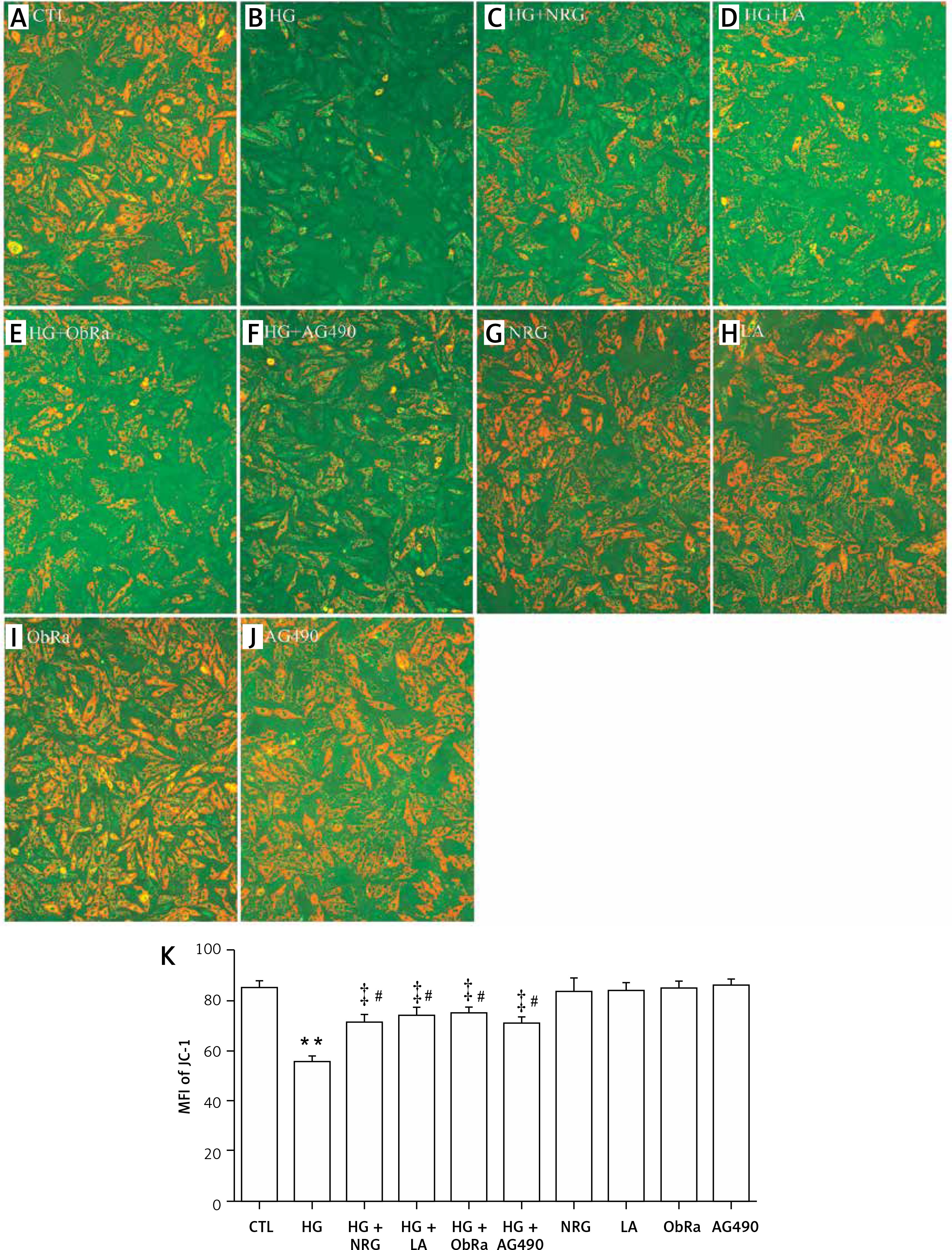
NRG, LA, ObRa, and AG490 improved MMP dissipation in HG-treated cardiac cells
As shown in Figure 8 B and K, the exposure of H9c2 cells to HG triggered mitochondrial damage, leading to MMP dissipation when compared to the control group (Figure 8 A). However, pretreatment with 80 μM NRG for 2 h significantly reduced MMP dissipation (Figures 8 C and K). Similarly, pretreatment with LA for 24 h (Figures 8 D and K), ObRa for 2 h (Figures 8 E and K), or AG490 for 30 min (Figures 8 F and K) significantly diminished MMP dissipation.
Figure 8
NRG, LA, ObRa, and AG490 ameliorated the dissipation of MMP in the HG-treated H9c2 cardiac cells. A–J – After the indicated treatment, MMP was assessed by JC-1 staining followed by photofluorography. A – Control group. B – Cells were treated with 35 mmol/l of glucose for 24 h. C – Cells were pretreated with 80 μmol/l of NRG for 2 h prior to exposure to HG for 24 h. D – Cells were pretreated with 50 ng/ml of LA for 24 h prior to exposure to HG. E – Cells were pretreated with 25 ng/ml of ObRa for 2 h before exposure to HG. F – Cells were pretreated with 10 μmol/l of AG490 for 30 min prior to exposure to HG. G – Cells were treated with 80 μmol/l of NRG for 2 h. H – Cells were pre-treated with 50 ng/ml of LA for 24 h. I – Cells were pretreated with 25 ng/ml of ObRa for 2 h. J – Cells were pre-treated with 10 μmol/l of AG490 for 30 min. K – Quantitative analysis of the MFI of JC-1 using ImageJ 1.47i software and the data in A–J
Data are shown as the mean ± SEM (n = 5). **P < 0.01, compared with the control group. ‡P < 0.01, compared with the HG-treated group. #P < 0.05, compared with the control group. NRG – naringin, LA – leptin antagonist, ObRa – leptin receptor antagonist, AG490 – an inhibitor of the JAK2/STAT3 pathway.
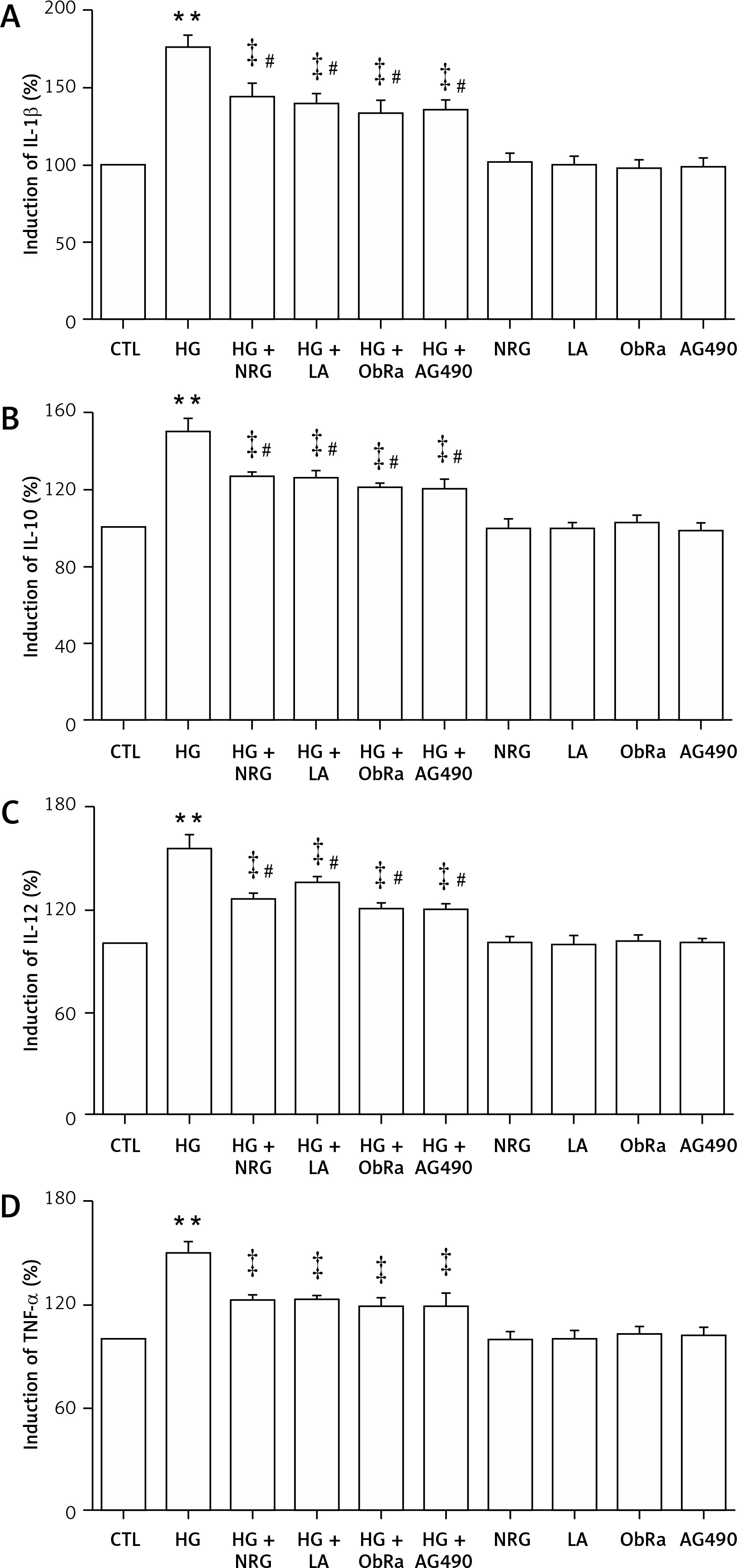
NRG, LA, ObRa, and AG490 attenuated the HG-induced production of pro-inflammatory cytokines in HG-treated cardiac cells
As shown in Figure 9, the production of interleukin (IL)-1β (Figure 9 A), IL-10 (Figure 9 B), IL-12 (Figure 9 C), and tumor necrosis factor (TNF)-α (Figure 9 D) was markedly increased in HG-induced H9c2 cells compared with the control group (p < 0.01). However, pretreatment with 80 μM NRG for 2 h before exposure to HG markedly reduced the secretion of IL-1β (Figure 9 A), IL-10 (Figure 9 B), IL-12 (Figure 9 C), and TNF-α (Figure 9 D). Similarly, pretreatment with LA for 24 h, ObRa for 2 h, or AG490 for 30 min prior to exposure to HG also diminished the production of these inflammatory cytokines (Figures 9 A–C).
Figure 9
NRG, LA, ObRa, and AG490 suppressed the HG-induced increase in production of pro-inflammatory cytokines in HG-treated H9c2 cardiac cells. H9c2 cells were divided into 10 groups (control group, HG group, HG + NRG group, HG + LA group, HG + ObRa group, HG + AG490 group, NRG group, LA group, ObRa group, and AG490 group). Cells were exposed to 35 mmol/l glucose for 24 h in the absence or presence of pretreatment with 80 μmol/l of NRG for 2 h, 50 ng/ml LA for 24 h, 25 ng/ml of ObRa for 2 h, or 10 μmol/l of AG490 for 30 min prior to exposure to HG. An enzyme-linked immunosorbent assay was performed to determine the levels of IL-1β, IL-10, IL-12, and TNF-α in cell supernatants. A–D – HG-induced increases in IL-1β, IL-10, IL-12, and TNF-α were suppressed by pretreatment with NRG, LA, ObRa, or AG490
Data are presented as the mean ± SEM (n = 5). **P < 0.01, compared with the control group. ‡P < 0.01, compared with the HG-treated group. #P < 0.05, compared with the control group. LA – leptin antagonist, ObRa – leptin receptor antagonist, AG490 – an inhibitor of the JAK2/STAT3 pathway.
Discussion
This study revealed several novel findings. First, in H9c2 cardiac cells, the leptin-JAK2/STAT3 signaling pathway is involved in HG-induced cell injuries and associated inflammatory reactions. Second, the JAK2/STAT3 pathway is a downstream signaling molecule of leptin. Third, NRG exerts cardioprotective effects against HG-induced injuries and inflammatory responses, which is consistent with our previous research [2–4]. Finally, the cardioprotective effects of NRG are associated with inhibition of the leptin-JAK2/STAT3 pathway.
Our data showed that the exposure of H9c2 cells to HG markedly induced injuries and inflammatory responses, in line with previous research [2–5]. In addition, these injuries and inflammatory responses were significantly alleviated by LA or ObRa, suggesting that leptin and its receptor were involved in HG-induced injuries and inflammatory responses [3, 24]. However, HG-induced H9c2 cell injuries and inflammation could not be completely suppressed by inhibition of the leptin pathway. These findings indicated that HG-induced H9c2 cell injuries and inflammatory responses are multifactorial and deserve further elucidation.
Notably, the JAK2/STAT3 pathway is wildly involved in the survival signaling pathway as well as cell proliferation, differentiation, growth, and apoptosis [25–28]. A recent study has demonstrated that N-acetylcysteine, a ROS scavenger, reduces myocardial ischemia reperfusion injury via upregulation of the JAK2/STAT3 pathway in diabetic animals [29]. In addition, it has been shown that the JAK2/STAT3 pathway mediates cardioprotection in streptozotocin-induced type 1 diabetic rats. However, the effects and mechanisms of the JAK2/STAT3 pathway on HG-induced cardiac cell injuries and inflammatory responses remain poorly understood. In order to explore these issues, we first investigated the roles of HG on the activation of the JAK2/STAT3 pathway in H9c2 cells. Our data showed that HG increased p-JAK2/p-STAT3 expression, indicating that HG could activate the JAK2/STAT3 pathway. Our finding reflected those of a previous report [30] but was in contrast to another study [19]. These discrepancies might be due to the different study protocols in terms of the stimulation methods used. Cardiomyocytes were treated with both HG and hypoxia/reoxygenation injury in the previous study versus only HG in the current study. In addition, our data indicated that H9c2 cardiac cells pretreated with AG490 significantly inhibited a variety of HG-stimulated cellular injuries, including apoptosis, cytotoxicity, mitochondrial damage, and oxidative stress. These results suggest that JAK2/STAT3 activation is involved in multiple HG-stimulated injuries. Our results are supported by several recent reports [31–33]. Previous research has found that AG490 exerts anti-inflammatory effects [34, 35]. Zhang et al. found that inhibition of the JAK2/STAT3 pathway was involved in anti-inflammatory effects against septic shock-triggered myocardial injuries [32]. Therefore, we postulated that the JAK2/STAT3 pathway may be associated with myocardial inflammation. In the present study, we further observed the roles of JAK2/STAT3 activation on HG-induced inflammatory responses. Similar to previous reports [4, 5], we found that the exposure of H9c2 cells to HG promoted inflammatory responses, as indicated by the upregulation of inflammatory cytokines including IL-1, IL-12, and TNF-α. However, the increased levels of inflammatory cytokines were decreased by AG490, suggesting that the JAK2/STAT3 pathway is involved in the HG-induced production of these pro-inflammatory factors. These data provide novel evidence to support the notion that activation of the JAK2/STAT3 pathway contributes to HG-induced injuries and inflammation in H9c2 cells.
A novel finding of our research was the identification of an association between leptin and the JAK2/STAT3 pathway in the HG-induced cardiac cells injury model. A previous paper demonstrated that leptin exerts various physiological functions by activating the JAK2/STAT3 pathway [16]. However, the relationship between leptin and the JAK2/STAT3 pathway is unknown under diabetic conditions. Our study found that the exposure of H9c2 cells to HG increased the expression levels of leptin and ObR and activated the JAK2/STAT3 pathway (up-regulation of p-JAK2 and p-STAT3). We also observed that pretreatment with LA or ObRa markedly suppressed the activation of JAK2/STAT3 (down-regulation of p-JAK2 and p-STAT), indicating that JAK2/STAT3 is a downstream molecular of leptin. Furthermore, pretreatment with LA, ObRa, or AG490 significantly antagonized HG-induced injury and inflammatory responses in H9c2cells, suggesting that the leptin-JAK2/STAT3 pathway mediated the HG-induced injuries and inflammatory responses in H9c2 cells. However, NRG, LA, ObRa, and AG490 were unable to completely inhibit HG-induced apoptosis, cytotoxicity, mitochondrial damage, or oxidative stress. We are uncertain about the underlying mechanisms. We believe that it will be helpful to evaluate other activating signaling cascades such as ERK1/2, JNK, P38 MAPK, PKC, and NF-κB in future studies to elucidate whether other signaling pathways are also involved.
Another novel finding of this study was that we discovered that the cardioprotective effects of NRG on HG-induced injuries and inflammatory responses were mediated by inhibition of the leptin-JAK2/STAT3 pathway. Our results showed that NRG exerted cardioprotective effects by inhibiting HG-induced injuries and inflammatory responses, which was consistent with our previous reports [2–4]. In addition, NRG inhibited the leptin pathway [3] and JAK2/STAT3 pathway [22]. We further demonstrated the effects of NRG on leptin and JAK2/STAT3 inhibition. Pretreatment with NRG significantly diminished the activation of the leptin pathway (downregulation of the expression levels of leptin and ObR) and JAK2/STAT3 activation (downregulation of p-JAK2 and p-STAT). Our data demonstrated that NRG suppressed the activation of the leptin-JAK2/STAT3 pathway in HG-induced H9c2 cardiac cells. Furthermore, the inhibitory effects of NRG on the leptin-JAK2/STAT3 pathway contributed to the cardioprotective effects against HG-induced H9c2 cells injuries and inflammatory responses.
The limitation of our current study was that we could not demonstrate a causal relationship between the JAK/STAT3 pathway and leptin. However, we considered that the findings from our study might, at least partially, have provided insights into the association between the JAK/STAT3 signaling pathway and leptin in cardiomyocytes. In future studies, the addition of recombinant leptin protein to cardiomyocytes in vitro and measurements of the changes in p-JAK2, p-STAT3, total (t)-JAK2, and t-STAT3 protein expression would demonstrate whether there is a causal relationship between JAK/STAT3 expression and leptin. Moreover, the western blot analysis of p-JAK2 and p-STAT3 did reveal the variations among different sets of the experiments, and the discrepancies may be due to the semi-quantitative method of densimetric analysis as well as the variations in the processing procedures among different sets of western blot analysis. As such, we should be cautious when interpreting our results and further studies may employ the immunohistochemically analysis to consolidate our data in western blot analysis.
In conclusion, this study provides novel evidence that the leptin-JAK2/STAT3 pathway contributes to HG-induced cardiac cell injuries and inflammation and NRG exerts cardioprotective effects against HG-induced cardiac cell injuries and inflammatory responses, and the effects may be related to the modulation of the leptin-JAK2/STAT3 signaling pathway. In line with our current findings, further studies should be conducted to develop and evaluate the novel therapeutic methods for the prevention and treatment of hyperglycemia-associated cardiac cell injuries and inflammation in the future.