Introduction
Ischemic cerebrovascular events cause significant morbidity and mortality [1]. Ischemia results in energy failure followed by loss of cell membrane pump functions, disruption of ion homeostasis, intracellular accumulation of Na+ and Ca2+, acidosis, glutamate excitotoxicity, free radical damage, accumulation of arachidonate oxidation products, glial activation, breakdown of the blood-brain barrier, leukocyte infiltration, and activation of caspases [2–4]. Cell death may occur through both apoptosis and necrosis [5, 6].
The only current clinical measure of preventing persistent brain damage following ischemia is immediate reperfusion, comprising resuscitation within seconds to minutes in global ischemia and thrombolysis within minutes to a few hours in focal ischemia. Hypothermia is also widely used as an adjunctive measure of treatment. However, both are limited by the difficulty of administration, shortness of time, inefficiency, and unwanted side effects. Furthermore, reperfusion is not singlehandedly life-saving, and even the reverse may be true when the severity of brain damage increases, leading to the development of ischemia/reperfusion (I/R) damage [7]. This situation has led to the search for neuroprotective drugs. Unfortunately, among more than 50 different drugs evaluated for neuroprotection, none yet has been proven to be sufficient for clinical use [4, 8].
RS100642 (1-(2’,6’-dimethyl-phenoxy)-2-ethylaminopropane hydrochloride) is a mexiletine analogue with less cardiotoxicity and neurotoxicity than those observed with mexiletine [9]. It blocks sodium channels with binding properties similar to mexiletine (pKi = 5.09 μM) but exhibits a more potent use-dependent reduction in compound action potentials as determined by rat vagus nerve preparations [10]. RS100642 has been examined in in vivo models of focal cerebral ischemia and was found to be beneficial in terms of neuroprotection not only in reducing the infarct size but also regulating the expression of Nav1.1 channel protein, reflecting that the effect results directly from Na+ channel blockade [11, 12]. However, RS100642 has not yet been tried clinically, nor has it been studied in in vivo global brain ischemia experimentally.
Therefore, this study aimed to evaluate the potential neuroprotective effects of RS100642 in global transient ischemia.
Material and methods
Animals and surgical procedure
Twenty-seven male Wistar Albino rats aged 7-9 months and weighing 229–308 g were obtained from the Experimental Animal Breeding and Research Center of Inonu University. Prior to the study, all animals were acclimatized to the housing conditions for 1 week on a 12-hour light/dark cycle at 18–22°C room temperature in cages sized 40 × 21 × 15 cm with 5 rats in each cage and fed with ad libitum laboratory chow and tap water. The experimental protocol was approved by the Ethics Committee for Animal Experiments of Inonu University. The rats were divided into 3 groups, namely control (n = 7), ischemia (n = 10) and RS100642 (n = 10). RS100642 was obtained from Roche Bioscience (Palo Alto, CA, USA). The compound was dissolved in a vehicle of distilled, deionized water immediately prior to testing.
Global ischemia was performed according to the 4-vessel occlusion method defined by Pulsinelli and Brierley [13]. Briefly, on the 1st day, all rats were anesthetized with ketamine + xylazine intraperitoneal (i.p.), followed by midsagittal incisions on the backs of their necks behind the occipital bones, dissection of the paravertebral muscles, and electrocauterization of both vertebral arteries. After electrocauterization, the rats were placed on their backs, and ventral cervical midsagittal incisions were made, followed by placement of loose silk sutures around both carotids, and suturing of the incisions. On the 2nd day, the rats were briefly anesthetized with ether. In the controls, only the sutures were removed and incisions were re-sutured, inducing no global brain ischemia. On the other hand, the rats in both ischemia and RS100642 groups had their sutures removed, both their carotids concomitantly were clasped tightly for 5 min, and the clasps were then removed for reperfusion. After 30 min of reperfusion, the RS100642 group rats received 1 mg/kg intravenous (i.v.) RS100642 at a volume of 1 ml/kg, while the ischemia group rats received i.v. isotonic saline at the same volume. The rats in both ischemia and RS100642 groups then had their incisions re-sutured and were left to recover. No specific premedication other than anesthesia was given prior to surgery, and paracetamol 300 mg/kg i.p. was administered postsurgically for analgesia. Three out of ten rats in the ischemia group and one out of seven rats in the control group were excluded from the study due to peri-operative mortality. Therefore, the investigations were conducted in 6 rats in the controls, 7 rats in the ischemia group, and 10 rats in the RS100642 group.
At the 6th hour following the procedure, all rats were sacrificed via decapitation under deep ether anesthesia. The brains were removed, the left hemispheres being preserved for 24 h at –40°C for biochemical investigations, and the right hemispheres being preserved for 24 h in 10% formalin for histopathological examinations.
Biochemical measurements
The left hemispheres of the brain samples were promptly dissected and perfused with 50 mM (pH: 7.4) cold phosphate buffer saline solution (PBS). Samples were homogenized in 1/5 (w/v) PBS. The homogenate was sonicated three times for 30 s with intervals and centrifuged at 20,000 × g for 15 min. Supernatants were separated and kept at –80°C until other biochemical measurements were performed.
The protein concentrations were determined according to the method of Lowry et al. [14], the malondialdehyde (MDA) levels were determined according to the method of Mihara and Uchiyama [15], the superoxide dismutase (SOD) activity was determined using the method of McCord and Fridovich [16], the catalase (CAT) activity was determined with the method of Luck [17], and the glutathione peroxidase (GPx) activity was measured with the method of Lawrence and Burk [18].
The activities of caspases-3 and -8 and tumor necrosis factor (TNF)-α levels were measured using ELISA kits (BioVision Research Product, CA, USA and eBioscience Research Kits, CA, USA, respectively) according to the manufacturer’s instructions.
Histopathological evaluations
The brain samples were placed in 10% buffered formalin and embedded in paraffin. Sections of brain tissues were cut at 6 μm and mounted on slides and stained with hematoxylin–eosin (H + E) and immunohistochemically with the Rabbit Polyclonal Antibody to Active Caspase-3 (Ab4051) Kit. The Caspase-3 Kit was used as recommended by the manufacturer (Abcam, Cambridge, United Kingdom). All sections were systematically examined with a Leica DFC 280 light microscope and analyzed in the Leica Q Win Plus V3 Image Analysis System (Leica Micros Imaging Solutions Ltd.; Cambridge, UK).
We applied modified criteria developed by Grafe et al. and Radovsky et al., who outlined light microscopic structural features of neurons and brain tissues [19, 20]. By light microscopy, necrotic (ischemic) neurons can be identified as exhibiting nuclear pyknosis and cytoplasmic eosinophilia. Other cellular alterations such as hyperchromatic indented or sharply scalloped nucleus and swollen neurons were also included in the neuronal counts.
Histopathological injury (mononuclear, polymorphonuclear, macrophages infiltrate and hemorrhage and edema (cells swell up) and gliosis) was scored in brain sections according to the following semi-quantitative scale: normal = 0, mild injury = 1 (infiltration, hemorrhage, edema and gliosis area < 10% of total brain cortex area), moderate injury = 2 (infiltration, hemorrhage, edema and gliosis area 11% to 20% of total brain cortex area), severe injury = 3 (infiltration, hemorrhage, edema and gliosis area 21% to 40% of total brain cortex area); and complete cavitary infarct = 4 (infiltration, hemorrhage, edema and gliosis area > 41% of total brain cortex area). Necrotic (ischemic) neurons in affected neuronal populations were counted without regard for the total number of neurons in the region, and put into five ranked categories as follows: 0 necrotic neurons = 0; 1–15 = (+1); 16–30 = (+2); 31–50 = (+3); and > 50 = (+4) randomly at 20 different areas in the brain cortex with the 20× objective.
The H-Score method was used to score the degree of caspase-3 positive immunoreactivity of neurons and glial cells in total sections’ area of the brain cortex. This immunohistochemical semi-quantitative method consists of the percentages of positively stained cells multiplied by a weighted intensity of staining: H Score = Pi (i + 1), where Pi is the percentage of stained cells in each intensity category (0–100%), and i is the intensity indicating weak (i = 1), moderate (i = 2) or strong staining (i = 3) [21].
Statistical analysis
Statistical analysis was performed using SPSS for Windows 15.0 software (SPSS Inc., IL, USA). For analysis of biochemical parameters, comparison among groups was made with the analysis of variance (ANOVA) test, while post hoc intergroup comparisons were performed with the Tukey HSD test. P < 0.05 was regarded as statistically significant.
Results
Evaluation of biochemical parameters
Oxidative stress
The definitive statistical data regarding biochemical parameters are displayed in Table I. Post hoc comparison of the groups against each other with the Tukey HSD test revealed a significant decrease in the SOD activities in the ischemia group as compared with both the RS100642 group and the controls (p < 0.05). On the other hand, SOD activities did not differ significantly between the RS100642 and the control groups (p > 0.05). Also, MDA level in the RS100642 group differed significantly compared to that of the ischemia group. Contrastingly, the CAT and GPx activities were significantly lower in both ischemia and RS100642 groups as compared with the controls (p < 0.05), while the RS100642 group was not significantly different from the ischemia group with regard to both of these parameters (p > 0.05).
Table I
Results are displayed as mean ± SD. There was significantly lower SOD activity in the ischemia group compared to the control group and a significantly higher value in the RS100642 group compared to the ischemia group. Also, a significantly lower MDA level was found in the RS100642 group compared to the ischemia group. On the other hand, the GPx and CAT activities of both RS100642 and ischemia groups were significantly lower than those of the controls
| Groups (n) | CAT [U/mg protein] | SOD [U/mg protein] | GPx [U/mg protein] | MDA [nmol/mg protein] |
|---|---|---|---|---|
| Control (6) | 636.15 ±133.20 | 5.52 ±0.47 | 1.65 ±0.33 | 20.79 ±2.35 |
| Ischemia (7) | 473.31 ±138.02* | 4.74 ±0.22* | 1.04 ±0.14* | 24.84 ±1.98 |
| RS100642 (10) | 457.99 ±57.85* | 5.36 ±0.55# | 1.05 ±0.15* | 19.35 ±4.25# |
Apoptosis
Caspase-3 and -8 activities and TNF-α levels are displayed in Table II. Post hoc comparison of the groups against each other with the Tukey HSD test revealed significantly higher caspase-3 activity in the ischemia group as compared with the RS100642 and control groups (p < 0.05). On the other hand, no significant difference was found among the ischemia, control and RS100642 groups with regard to caspase-8 and TNF-α levels (p > 0.05).
Table II
Results are displayed as mean ± SD. There was significantly higher caspase-3 activity in the ischemia group as compared with the RS100642 and control groups, while the caspase-3 activities of RS100642 and control groups were similar. Caspase-8 activities and TNF- α levels did not differ significantly between groups
| Groups (n) | Caspase-3 [RFU/mg protein] | Caspase-8 [RFU/mg protein] | TNF-α [pg/ml] |
|---|---|---|---|
| Control (6) | 14.79 ±1.51 | 53.04 ±13.80 | 4.86 ±1.07 |
| Ischemia (7) | 19.91 ±2.43* | 65.74 ±19.18 | 5.62 ±1.49 |
| RS100642 (10) | 16.84 ±2.83# | 65.21 ±9.24 | 4.35 ±1.32 |
Evaluation of histopathological findings
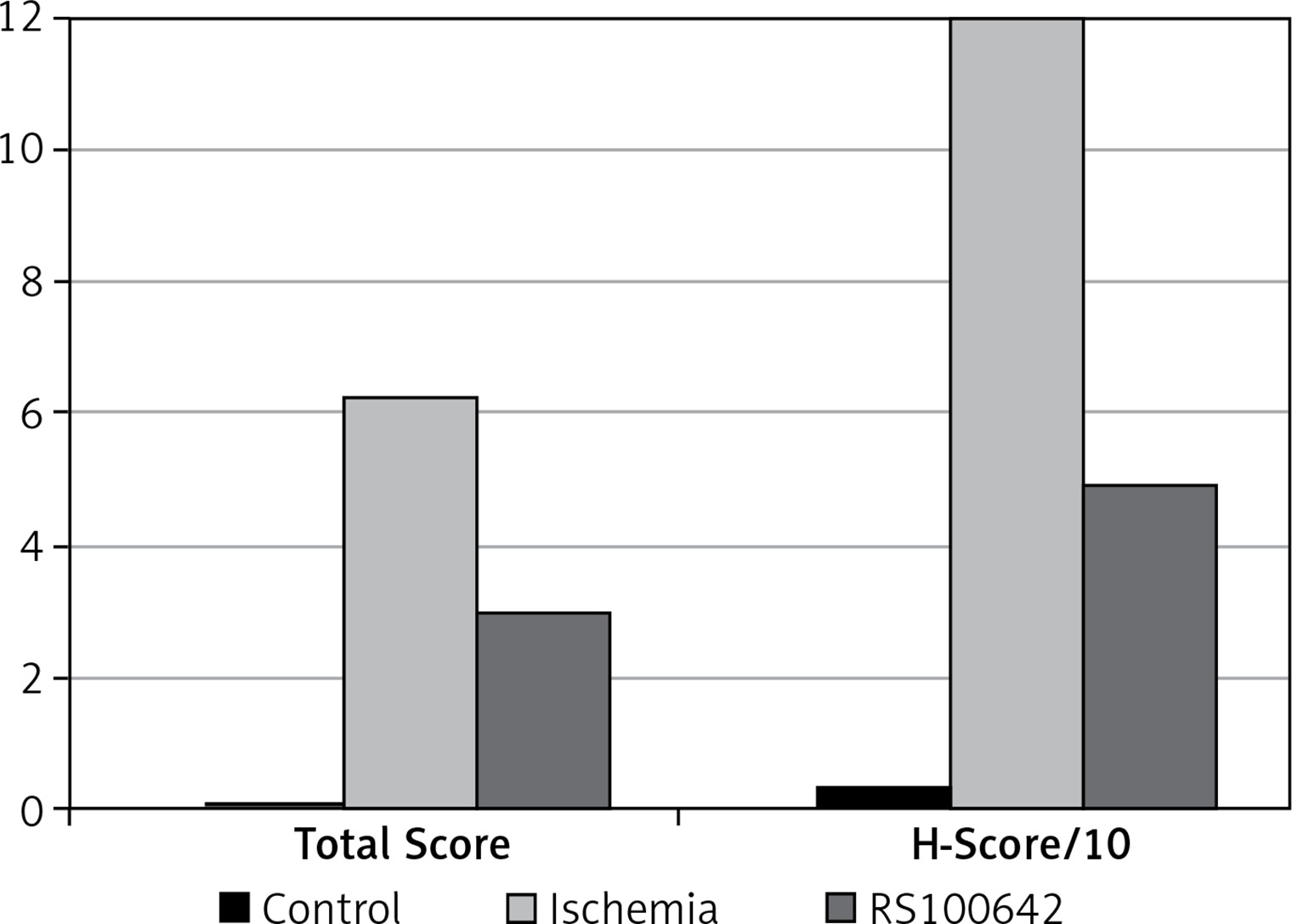
The samples of the control group were normal in histological appearance (Figures 1 A, B). The ratio of ischemic to healthy neurons, neuronal swelling, tissue edema, and other signs of histopathological injury including hemorrhage and infiltration (Figure 2 A) and caspase-3 positive immunoreactivity (Figure 2 B) in the ischemia group were significantly higher than in the control and RS100642 groups. The number of ischemic neurons, the severity of the histopathological injury and caspase-3 positive immunoreactivity were significantly lower in the RS100642 group (Figures 3 A, B) as compared with the ischemia group. The total score and H-score values in each group are shown in Figure 4.
Figure 1
Control group: A – the controls with the normal histological appearance of brain tissue. H + E, 40×, B – absence of caspase-3 positive immunoreactivity, caspase-3, 40×
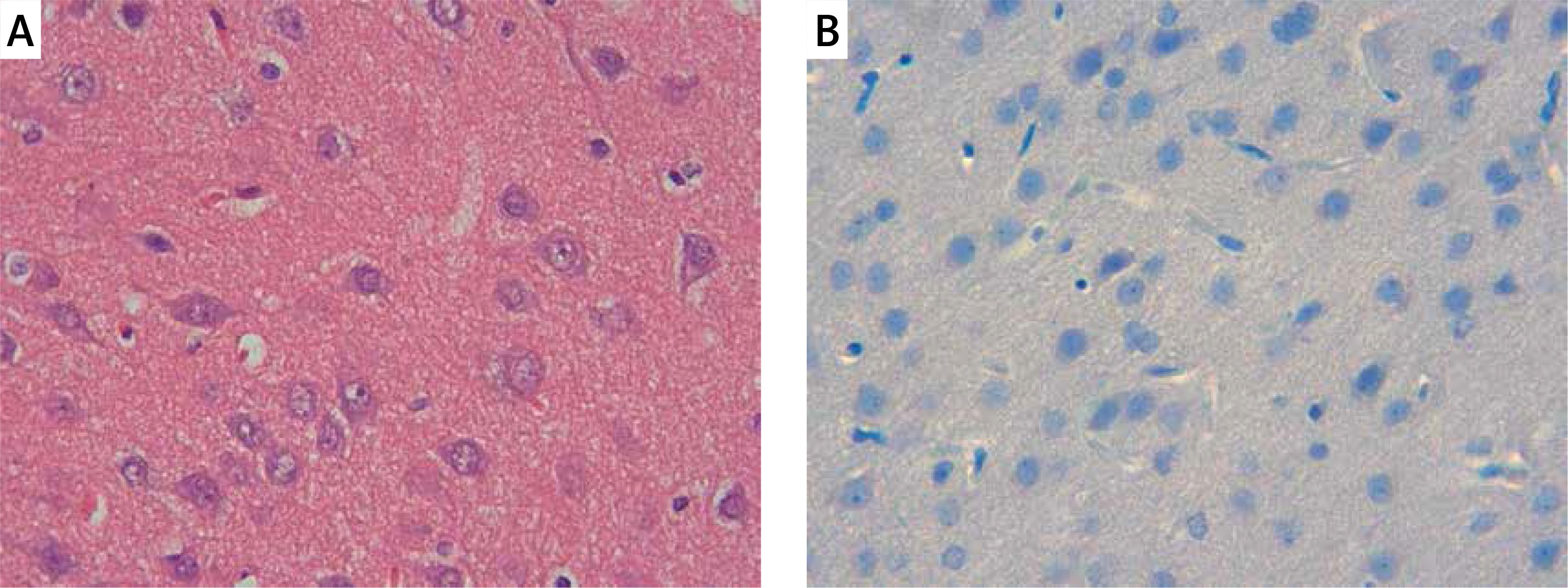
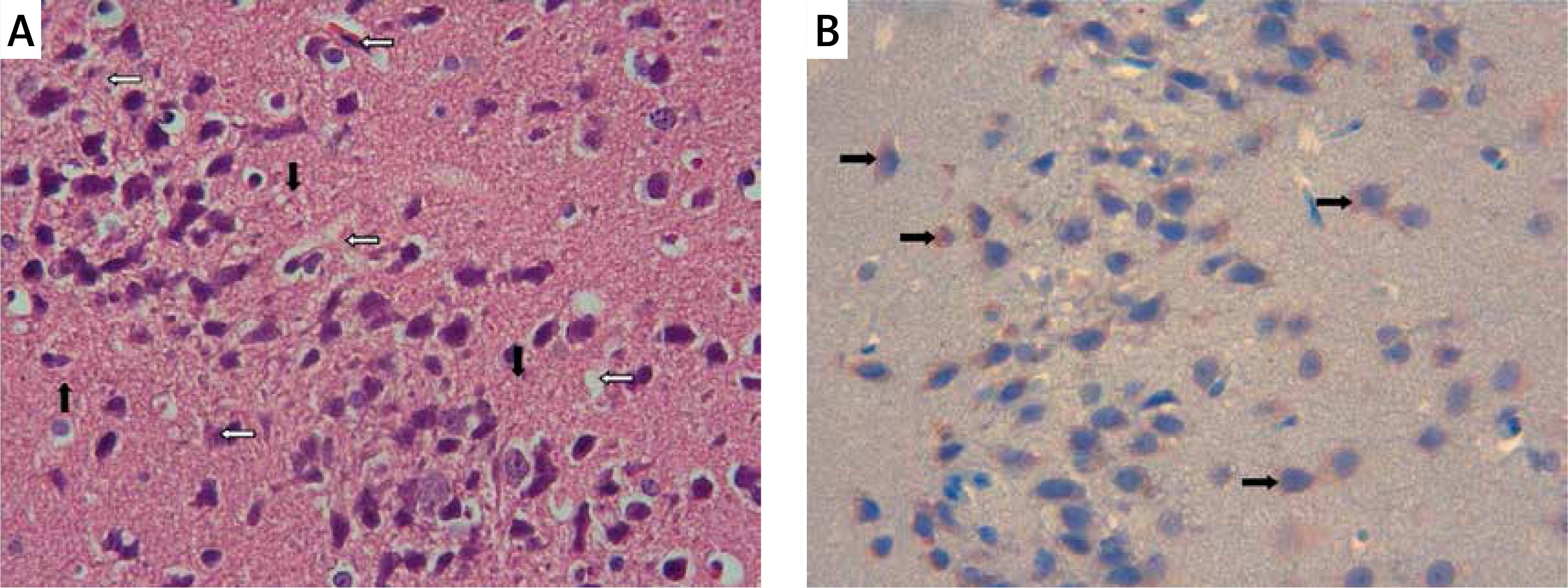
Figure 2
Ischemia group: A – necrotic (white arrows) and swollen neurons (black arrows). H + E, 40×, B – prominent caspase-3 positive immunoreactivity (black arrows). Caspase-3, 40×
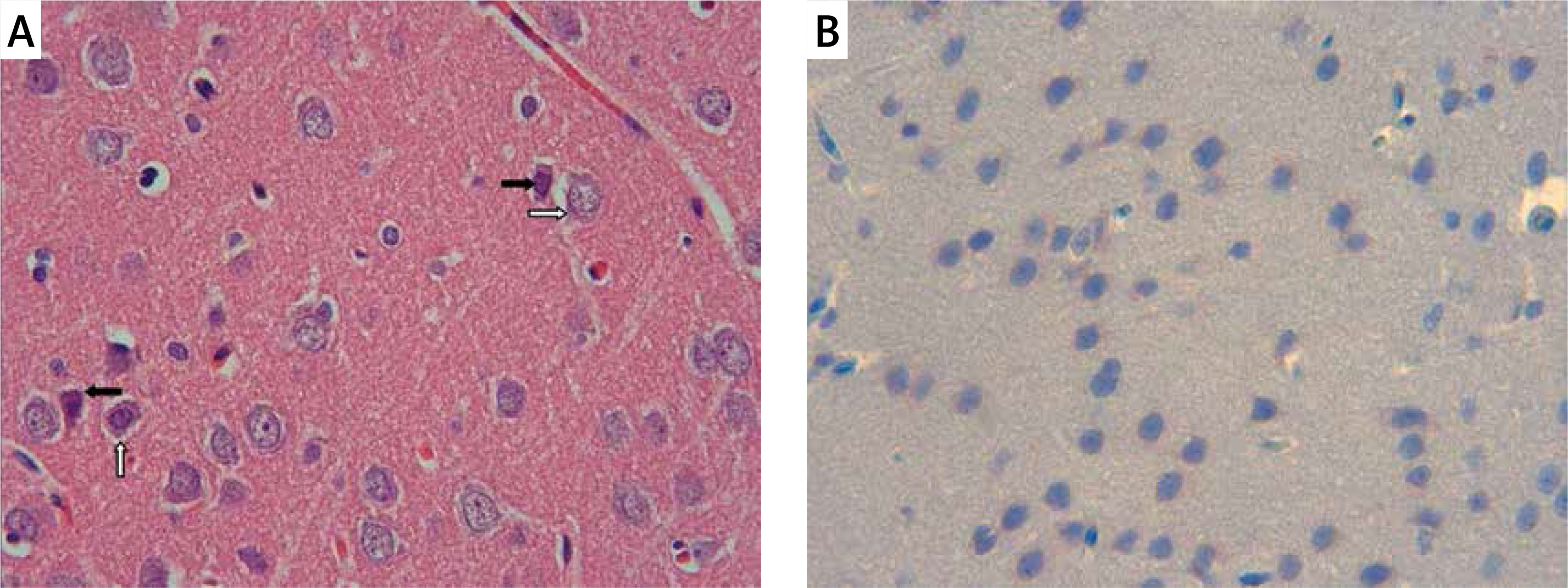
Discussion
We have demonstrated a promising neuroprotective effect of RS100642 against I/R injury in global ischemia of 5 min followed by reperfusion for 30 min, reflected strongly in the SOD activities, and partially in caspase-3 levels and histopathology. To date, RS100642 has been investigated in focal ischemia scarcely, and this is the first study of this agent in global ischemia. Global cerebral ischemia with 4-vessel blockade has been induced in several studies via clamping the carotids from 10 to 30 min [22, 23]. Cell death has been shown to take place in minutes after the initiation of global ischemia, and this is approximately 5 min in most sensitive parts of the central nervous system [24]. Therefore, we clasped both carotids concomitantly for 5 min to produce brain injury. We sacrificed the rats at the 6th h, as this duration is adequate for superoxide radical accumulation [25], disruption of the cell membrane, and disintegration of organelles in I/R damage [26]. We administered RS100642 at a dose of 1 mg/kg, the same dose that was found to be successfully neuroprotective in previous studies. Lower doses have also been tested in focal ischemia and were found to be less effective, while increasing the dose further was not beneficial [9].
Under physiological conditions, the brain is an organ with high O2 consumption and free oxygen radical production, but low antioxidant capacity. Due to the abundance of polyunsaturated fatty acids, it is particularly damaged by oxidative stress during I/R injury. Increased production of reactive oxygen species (ROS) due to I/R injury may easily overcome the antioxidant systems and result in oxidative damage to cellular components [25]. Increases in MDA levels and decreases in antioxidant enzyme activities in traumatic brain damage have been reported previously [27]. Therefore the MDA levels and SOD, CAT and GPx activities are reliable indicators of cerebral I/R damage. In our study, RS100642 suppressed early accumulation of MDA and enhanced the activity of SOD. The decrease in MDA levels reflects decreased lipid peroxidation, whereas the lower level of SOD activity reflects reduction in dismutation of superoxide radicals. However, RS100642 failed to prevent reduction of CAT and GPx levels significantly. Like SOD activity being used up against increasing O2 – levels, CAT and GPx activities are used up in order to reduce the increasing H2O2 levels [28, 29]. Neither CAT nor GPx activities differed significantly from those in the ischemia group with RS100642 administration, indicating that administration of RS100642 significantly protects from damage related to lipid peroxidation and O2 –, but not H2O2, which is regarded as the main radical responsible for necrosis [30, 31]. Necrosis occurs when intracellular ATP levels fall below 25% of the basal levels, and apoptosis commences if they remain higher [32]. Unless eliminated by CAT, H2O2 leads to failure of ATP synthesis via overloading of the cellular hexose monophosphate pathway for reduction of GS-SG and depletion of nicotinamide adenine dinucleotide (NAD+) but also directly suppresses ATP synthesis in the mitochondria [30], facilitates iron-mediated secondary free O2 radical formation, and peroxidizes cell lipids [33]. As mitochondria play a pivotal role in both Ca2+ metabolism and initiation of caspase cascade, reduction in ATP synthesis and increase in mitochondrial permeability lead to increased cytochrome c release and therefore apoptosis accompanied by poly (ADP-ribose) polymerase (PARP)- and c-Jun N-terminal kinase (JNK)-mediated necroptosis, a condition with features of both necrosis and apoptosis [2, 34, 35]. These effects of H2O2 may explain why necrosis persisted morphologically following RS100642 administration. The difference in parameters related to H2O2 and O2 – may arise from a free radical scavenging property of RS100642, which does not act on H2O2 and peroxynitrite. Although it has not yet been investigated whether RS100642 has free radical scavenging activity, mexiletine has been shown to be a potent free radical scavenger [36]. Further studies investigating whether RS100642 has free radical scavenging activity and whether the introduction of RS100642 in the company of a scavenger against H2O2 could be more beneficial are required to clarify these questions.
Caspase-dependent apoptosis may be initiated either by activation of caspase-8 by death receptors on the cell surface or by activation of caspase-3 through mitochondrial stress including cytochrome c (Cyt c) release [37]. Cyt c released from the mitochondria forms a complex with apoptosis activating factor-1 and pro-caspase-9, resulting in caspase-3 activation, cleavage of cellular substrates and cell death, as demonstrated in the delayed neuronal death following ischemia [38]. Translocation of Cyt c and activation of caspase-3 have been reported following global ischemia in gerbils [39], and following transient middle cerebral artery occlusion for 2 h, starting as early as the 30th min of reperfusion [40]. Accordingly, increased caspase-3 immunoreactivity in the cerebral cortex after cerebral I/R injury has been demonstrated histopathologically [41]. Likewise, histopathological images support our biochemical findings. While the brain specimens from control animals presented no histological alterations (Figures 1 A, B), those from animals in the ischemia group displayed necrotic and swollen neurons with partial edema as well as prominent caspase-3 positive immunoreactivity (Figures 2 A, B). The inhibition of histopathological changes observed in the RS100642 group is illustrated in Figures 3 A and B. It has been demonstrated that neurons derived from caspase-3-knockout mice are resistant to ischemic stress [42], and caspase inhibitors (z-DEVD–FMK or z-VAD–FMK) prevent ischemia-induced neuronal damage and improve neurological outcome [43]. Our results confirmed the increased caspase-3 activity and immunoreactivity following I/R injury in rats and demonstrated a beneficial effect of RS100642. The second pathway of apoptosis involves activation of caspase-8 through death receptors located on the cell surface. The death receptors belong to the tumor necrosis factor (TNF) superfamily, and the best-characterized ones are CD95 (or Fas), TNF receptor-1 and TNF-related apoptosis-inducing ligand (TRAIL) receptors DR4 and DR5. Cytotoxic effects of high levels of TNF-α are mediated either by its inflammatory action or binding to TNF receptor 1 (TNFR1)-type receptors on dopaminergic neurons leading to activation of caspase-8-mediated apoptosis [44]. TNF-α can also induce the activation of inducible nitric oxide synthase (iNOS), amplifying the inflammatory response and leading to the production of more cytokines [45]. We observed considerable but statistically insignificant increases in TNF-α and caspase-8 levels in the ischemia group as compared with the controls, while these increases were particularly inhibited in the group with Na+ channel blockade. We, therefore, propose that apoptosis develops following global brain ischemia mainly by caspase-3 activation through the mitochondrial pathway rather than activation of death receptors, and can be prevented by RS100642 administration.
In conclusion, our results suggest that Na+ channel blockade by RS100642 has remarkable neuroprotective effects following global brain ischemia. In this context, RS100642 administration at a dose of 1 mg/kg following 5 min of global brain ischemia and 30 min of reperfusion significantly improves the MDA level and SOD and caspase-3 activities, all indicating free oxygen radical damage and apoptosis activation in the brain. However, further studies towards revealing the complete molecular mechanism underlying this neuroprotection, and determining the optimal effective dose and time of administration, and potential benefits of co-administration of RS100642 with other drugs, remain to be conducted.