Introduction
Hereditary angioedema (HAE) encompasses a group of rare diseases characterized by recurrent subcutaneous or submucosal long-lasting swellings (24 to 96 h when untreated) that commonly involve the extremities, the face, the genitals, the bowel and the upper airways [1]. Potentially, the occurrence of events such as laryngeal attacks or, rarely, hypovolemic shock is life-threatening [2, 3]. The most frequent disease type is caused by quantitative or functional deficiency of the serine protease inhibitor C1-esterase inhibitor (C1-INH) protein, due to mutations in the gene SERPING1 [1]. The resulting C1-INH-HAE is defined as type I or type II and it affects the complement, contact and fibrinolytic cascades, leading to uncontrolled production of vasoactive mediators, among which the release of bradykinin (BK) is crucial [4, 5]. BK is also the major mediator in the HAE subtype defined by normal C1-INH function [6]. Mutations in the F12 gene have been described in a large proportion of families affected by this subtype of HAE, named FXII-HAE [7, 8].
Oxidative stress is the result of an imbalance between endogenous production of free reactive oxygen species (ROS) and reduced effectiveness of antioxidant defense mechanisms. This imbalance can worsen inflammation and injury conditions by enhancing release of pro-inflammatory cytokines and altering enzymatic function [9]. Oxidative stress occurs in many inflammatory, neoplastic and immunologic diseases, including asthma, allergic rhinitis, obesity and leukemia [9–11].
Advanced oxidation protein products (AOPPs) and advanced glycation end products (AGEs) are a family of compounds generated in body fluids from oxidation of macromolecules, including proteins, used as markers of oxidative stress and inflammation in several diseases and their complications [11–15]. The AOPPs are protein derivatives, predominantly albumin, and its aggregates mainly formed by chlorinated oxidants. The AGEs, e.g., pentosidine and carboxymethyllysine, are formed via autoxidation of sugars as well as other glycation intermediates and via lipoperoxidation of polyunsaturated fatty acids.
Kinin vasoactive peptides participate in inflammatory processes through the binding and activation of two G-protein-coupled receptors (R): B1R, inducible by proinflammatory cytokines and oxidative stress via the transcriptional nuclear factor κB (NF-κB), and B2R, which is expressed constitutively and increases vascular permeability [16]. B1R is a potent activator of inducible nitric oxide and NADPH oxidase, which are associated with vascular inflammation, increased permeability, and endothelial dysfunction [17]. However, there are no data available data on oxidative stress in patients with HAE.
This study aimed to assess the serum concentrations of AGEs and AOPPs in patients affected by HAE due to C1-INH deficiency and HAE with normal C1-INH.
Material and methods
Study subjects, laboratory tests and HAE diagnosis
We consecutively investigated patients from families with C1-INH-HAE and families diagnosed with FXII-HAE. Patients were recruited at the Unit of Internal Medicine, Allergy and Clinical Immunology (University of Cagliari, Italy). We studied 19 C1-INH-HAE patients, 15 FXII-HAE patients and 21 healthy controls. The blood samples were taken during the remission state, which was defined as asymptomatic for at least 15 days before sampling. Furthermore, sera were collected prior to the prescription of drugs such as tranexamic acid, attenuated androgens or long-term prophylaxis with C1-INH.
Exclusion criteria at the moment of blood sampling were: the presence of concurrent diseases or conditions and the intake of drugs and dietary supplements known to interfere with oxidative stress marker levels.
The diagnosis of C1-INH-HAE and of FXII-HAE was established on the basis of complete clinical history and laboratory tests, according to published criteria [1]. In both groups, affected asymptomatic subjects were identified through familial screening.
Plasma levels of C1-INH antigen were assayed by radial immunodiffusion (NOR Partigen C1-INH, Siemens Healthcare Diagnostics, Marburg, Germany), C4 antigen by nephelometry, and C1-INH activity in plasma using a chromogenic assay (Technochrom C1-Inhibitor, Technoclone, Vienna, Austria). Quantitative D-dimer assay was performed by a latex immunoassay (Hemosil D-dimer HS, Instrumentation Laboratory SpA, Italy).
Based on the clinical history, the patients were previously evaluated as appropriate on the basis of known causes of angioedema, including the sequence of SERPING1 and F12 genes [18–20]. C1-INH-HAE and HAE-FXII severity scores were calculated respectively according to Gomez-Traseira et al. and Bygum et al. [21, 22].
The study was carried out after approval by the local ethics committee (protocol NP/2013/3226), in accordance with the Principles of the Declaration of Helsinki and its amendments; written informed consent for the study was obtained from all the participants.
Advanced glycation end products and advanced oxidation protein products
The AGEs and AOPPs were measured by spectrofluorimetric and spectrophotometric methods, respectively, as previously described [12, 23].
For AGE determination, serum was diluted 1 : 50 with phosphate-buffered saline (PBS; pH 7.4), and fluorescence intensity was recorded at maximum emission (~440 nm) upon excitation at 350 nm and expressed in arbitrary units (AU). The serum concentration of AGEs was normalized to the total protein amount determined by the Bradford assay and expressed in AU per gram of protein.
For AOPP determination, 200 μl of blood serum diluted 1 : 5 with PBS, 200 μl of chloramines T (0–100 mol/l) for calibration, or 200 μl of PBS as blank were applied on a microtiter plate. Ten μl of 1.16 M KI and 20 μl of acetic acid were added, and absorbance at 340 nm was measured immediately. The serum concentration of AOPPs was normalized to the total protein amount determined by the Bradford assay and expressed as chloramines nmol/mg of protein.
Each sample was analyzed in triplicate for both AGE and AOPP determination.
Statistical analysis
Statistical analysis was performed using SPSS 22 for Windows. Data were analyzed using non-parametric methods. Statistical significance was assessed by the Kruskal-Wallis H test, which does not assume a normal distribution of measured variables. Results of concentrations of AOPP and AGE levels are expressed as median (interquartile range). Post hoc analysis based on the Dunn-Bonferroni approach was performed and statistical significance was achieved when p < 0.05.
Results
The characteristics of the population investigated are summarized in Tables I–III.
Table I
Clinical features of healthy subjects and patients affected by HAE
| Parameter | Healthy subjects (n = 21) | C1-INH-HAE (n = 19) | FXII-HAE (n = 15) |
|---|---|---|---|
| Sex (M/F) | 6/15 | 7/12 | 3/12 |
| Age, average [years] | 37.5 | 49 | 38 |
| Range [years] | 18–54 | 16–81 | 10–66 |
Table II
Clinical and laboratory data of patients affected by FXII-HAE
Table III
Clinical and laboratory data of patients affected by C1-INH-HAE
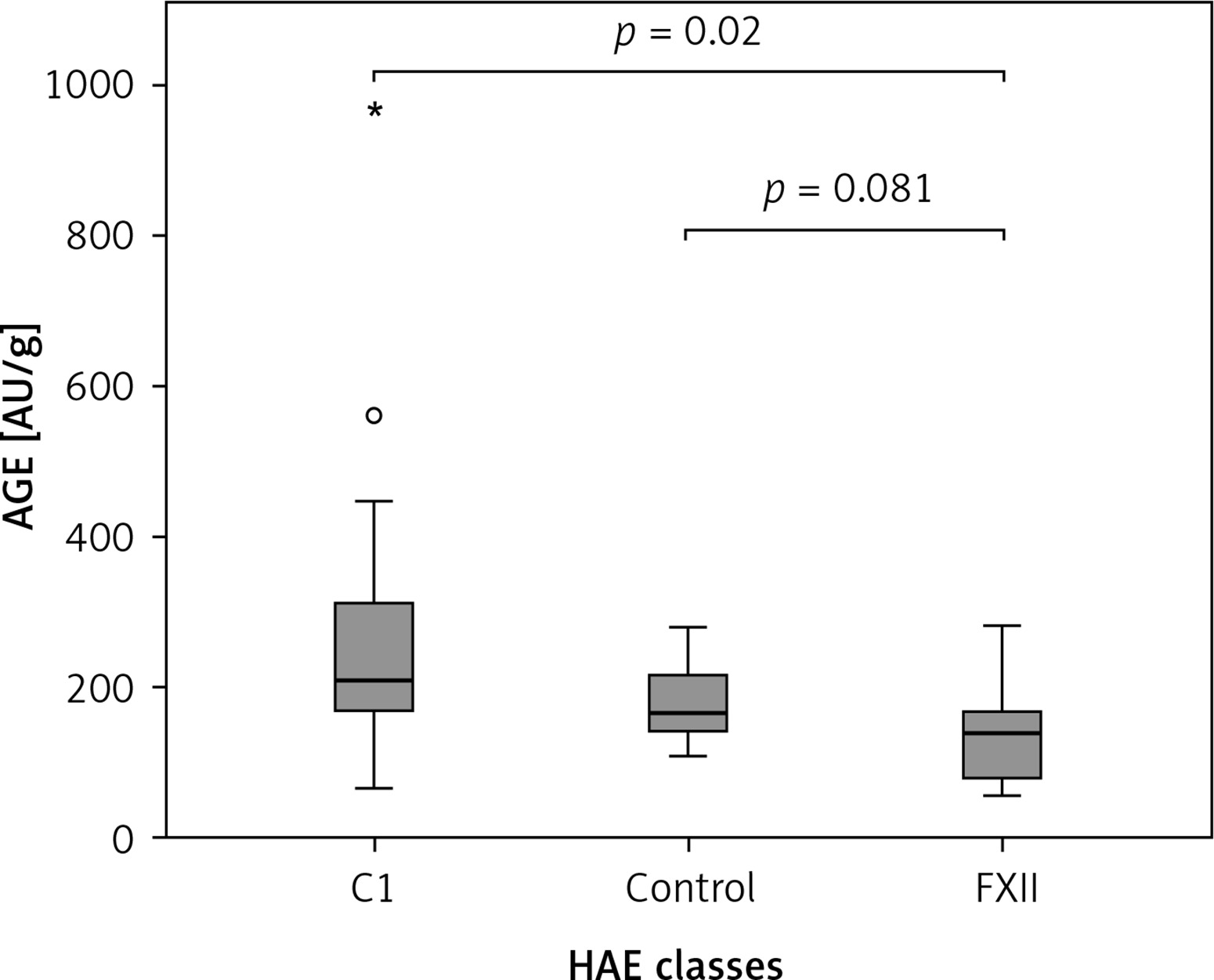
The Kruskal-Wallis H test showed that there was a statistically significant difference in AOPP (nmol/mg) between the three different groups, χ2(2) = 26.574, p = 0.001, with a mean rank AOPP value of 39.63 for C1-INH-HAE, 32.47 for FXII-HAE and 14.29 for the control group. Post hoc analysis of AOPP levels (Figure 1) showed that the control group had a concentration of 0.94 (0.36) nmol/mg, which was significantly lower than levels in the C1-INH-HAE group (1.68 (0.47) nmol/mg; p = 0.002) or FXII-HAE (1.50 (0.27) nmol/mg; p = 0.001). Also differences in AGE levels (AU/g) were statistically significant, χ2(2) = 9.18, p = 0.010, with a mean rank AGE value of 35.37 for C1-INH-HAE, 18.60 for FXII-HAE and 28.05 for the controls. Post hoc analysis of AGE levels (Figure 2) showed that the value in the FXII group (141.48 (89.59) AU/g) was lower and significantly different versus the C1-INH-HAE group (211.58 (151.05) AU/g; p = 0.02) and comparable with the control group (167.02 (78.76) AU/g; p = 0.081).
Figure 1
The AOPP (nmol/mg protein) concentration among HAE patients and healthy controls. Box plot illustrates median (IQR) AOPP levels in groups of individuals of different HAE classes (C1, FXII) and control group. The Mann-Whitney U test was used for paired comparisons; p-values and medians are indicated in the figure
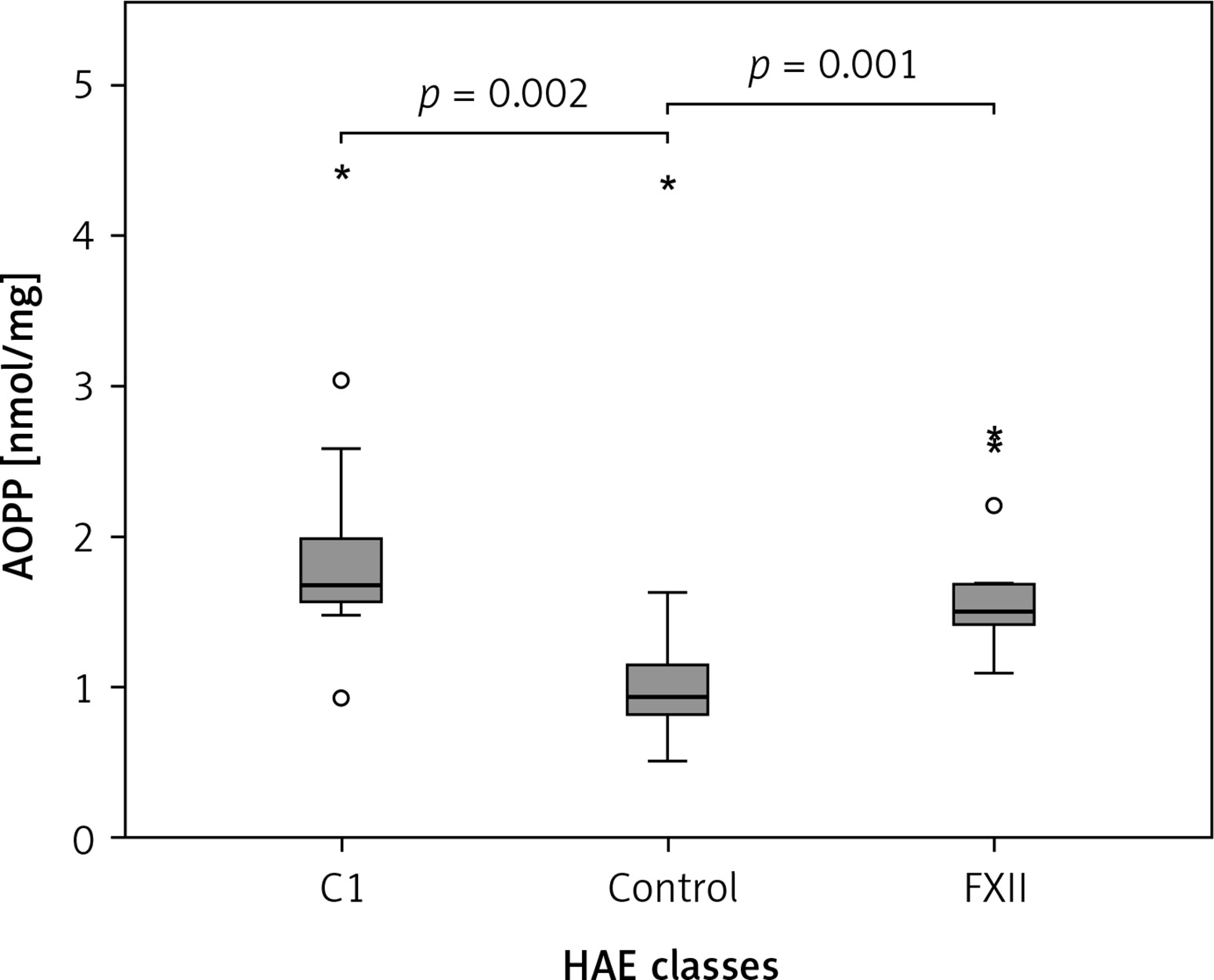
Figure 2
The AGE (AU/g protein) concentration among HAE patients and healthy controls. Box plot illustrates median (IQR) AGE levels in groups of individuals of different HAE classes (C1, FXII) and control group. The Bonferroni post hoc test was used for paired comparisons; p-values and medians are indicated in the figure
Discussion
There is increasing interest in identification of specific biomarkers, defined by the National Institutes of Health as “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention”. In particular, biomarkers of oxidative stress can be classified as molecules (DNA, lipids, proteins or carbohydrates) modified by interactions with ROS in the microenvironment; and molecules of the antioxidant system that change in response to increased redox stress. The AGEs and AOPPs can serve as densitometric markers of oxidative stress and inflammation in several diseases and their complications [10, 11, 13, 15, 23–25]. In fact, modified proteins may be, more efficiently than other biomarkers, employed to monitor disease progression and outcome, since proteins are, in general, key molecules that play the ultimate role in various structural and functional aspects of living organisms, and their activity and function are strictly dependent on structure, conformation and folding pattern. Thus, modification of conformation/structure of the polypeptide chain in conditions of oxidative stress/inflammation can lead to dysfunction/function, loss of proteins, and inhibition of their degradation (and, consequently, their accumulation) and also has a wide range of downstream functional consequences, causing subsequent cellular dysfunction, tissue damage, and disease onset/progression. Furthermore, as the techniques used to measure AGEs and AOPPs are simple, fast, and inexpensive, they may be applicable in daily routine laboratory practice for assessing and monitoring oxidative stress in critically ill and several other types of patients. These biomarkers, as well as other modified proteins, also have the further advantages of relative stability and consequent higher blood concentrations [14].
Our data indicate that both C1-INH-HAE and FXII-HAE are associated with a state of increased oxidative stress, as shown by the increased circulating levels of AOPPs and, in C1-INH-HAE only, of AGEs. Bradykinin generation and signaling appear to be important in HAE as well as angioedema due to ACE inhibitors [26], but they contribute to several other diseases, in particular anaphylaxis and mast-cell mediated allergic reactions [27, 28], as well as systemic mastocytosis [29], which is characterized by increased circulating AOPP levels.
On the other hand, serum AOPP levels were higher in C1-INH-HAE patients than in FXII-HAE patients, and AGE serum levels were higher only in C1-INH-HAE patients than in controls.
It has been proposed that FXII-HAE may be potentially more closely linked with BK and B2 receptors, and C1-INH-HAE with desArg9-BK and B1 receptors [8, 30, 31].
In fact, both the B1 and B2 receptors mediate vasodilation and permeability increase and, through their binding to the receptors, there is activation of G-proteins and downstream effectors including eNOS, PLA2 and PLC-β1 [17]. In general, the protective role of B2R in oxidative stress-mediated disorders of the kidney and heart is recognized [32], through eNOS induction and tonic NO production [33].
Increased eNOS levels in attack-free periods have been observed in C1-INH-HAE patients [34], although with evidence of an underlying endothelial dysfunction [35, 36]. In fact, bradykinin receptors may have dual beneficial and deleterious effects in vascular and inflammation physiopathology [17, 37]. Besides the B2R-like pathways, B1R leads to prolonged signaling and to stimulation of iNOS, which generates a much higher and prolonged output of NO with the potential to promote inflammatory responses [38, 39]. High levels of NO can react with superoxide anion to generate peroxynitrites and increase oxidative stress in mice [40].
Activation of B1R increases the oxidative stress through the activation of NADPH oxidase, resulting in increased superoxide anions in human cells and rat models [41, 42].
Our data might be linked to differences in bradykinin receptor signaling, resulting in a different pattern of the measured oxidative stress markers among HAE subgroups.
The BK formation may occur independently from factor XII and kallikrein, relying on the lectin pathway of the complement system and activation of MBL-associated serine protease (MASPs), which are also activated upon oxidative stress [43]. The potential impact of the measured markers of oxidative stress in HAE attacks through MASPs deserves further investigations. C1-INH-HAE is associated with dysregulation of complement, coagulation, and contact cascades [44], and high levels of procoagulant and fibrinolytic activity have been demonstrated in patients during acute attacks and remissions [45, 46]. Thus, the different behavior observed between C1-INH-HAE patients and FXII-HAE patients may be related to a different imbalance in the coagulation, contact and kinin pathways [47, 48]. The oxidation of fibrinogen plays a key role in the formation of AOPPs, which, in turn, may promote functional alterations in fibrinogen, increasing its procoagulant activity or modifying its direct effects on vascular tone [49, 50]. Furthermore, blood levels of AGEs are a biomarker known to be intimately involved in the pathophysiology of cardiovascular disease and appear to be directly associated with the onset of thrombotic risk; for example, we observed increased AGE serum levels in patients affected by essential thrombocythemia and who had experienced thrombotic events [15, 51].
The apparent potential thromboembolic risk in patients with C1-INH-HAE, with activation of fibrinolytic activity and coagulation as well as high levels of D-dimer, has not been confirmed by clinical observations and needs to be addressed with further studies [18, 45, 46].
Finally, FXII-HAE was previously named “estrogen-dependent” HAE; thus an explanation for the difference between the two HAE subgroups could be found in a different capability of estrogen to modulate vascular reactivity and inflammation, coagulation and platelet aggregation [31, 52, 53].
To our knowledge, this is the first study to investigate the role of oxidative stress in HAE, highlighting additional underlying mechanisms involved in this disease. Further studies are in progress to better characterize the effects of oxidative stress on different systems involved in HAE pathophysiology.