

Introduction
Chronic inflammation develops when the immune system is unable to clear a persistent insult. Unresolved chronic inflammation leads to disruption of tissue homeostasis, resulting in inflammatory diseases, including neurodegenerative diseases, autoimmune diseases, metabolic disorders and cancers [1, 2]. To prevent excessive immune stimulation, chronic inflammation induces immunosuppression, which was mediated primarily by myeloid-derived suppressor cells (MDSCs) [3].
MDSCs are a diverse population of immature myeloid cells that have the ability to suppress both adaptive and innate immunity through multiple mechanisms. In mice, M-MDSCs and PMN-MDSCs are defined as CD11b+ Gr-1+ Ly6Glow Ly6Chigh and CD11b+ Gr-1+ Ly6Ghigh Ly6Clow, respectively. Human MDSCs are defined according to expression of CD33, CD11b, HLA-DR, CD14, and CD15 [4]. In view of the plasticity and diversity of MDSCs, it was accepted that MDSCs could be identified as environmental sensors or adapters. When recruited to the site of inflammation, which is characterized by diversity of cytokines, chemokines and pro-inflammatory mediators, MDSCs would acquire different suppressive functions by the mechanism of changing their cell fate, surface receptors, metabolism pathway and suppressive activities [5, 6].
In chronic inflammatory disorders, stimulatory molecules that are released by damaged cell/tissue can activate myeloid cells by signaling through pattern recognition receptor (PRRs) such as Toll-like receptors (TLRs) [7, 8]. TLRs are a group of Class I transmembrane proteins with 11 members in humans and 13 in mice [9]. TLRs are expressed by immune cells, endothelial cells, and epithelial cells and their expression is regulated in response to the environment milieu such as pathogens, cytokines, and environmental stress [10]. TLRs can recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), including a broad range of microbial and non-microbial host-derived endogenous ligands or self-molecules, and thus contribute to both host defense and the development of chronic sterile inflammatory diseases [7]. Recent reports have suggested that the survival, differentiation, and suppressive activity of MDSCs are influenced by TLR signaling [11]. Among the TLRs, TLR2 could be activated by a vast number and diversity of TLR2 ligands [12].
Research on cancer, infection and autoimmunity demonstrated that TLR2 signaling promoted the accumulation and activation of MDSCs via different pathways [3, 13-17]. However, no indication of how TLR2 regulated the immunosuppressive mechanism of MDSCs in the context of sterile chronic inflammation excluding cancer and infections was provided. Moreover, it has also been demonstrated that TLR1/2 agonists decreased the MDSC population and dampened the suppressive activity of MDSCs by inducing tumoricidal M1-type macrophage characteristics in MDSCs [18-20]. Therefore the regulatory effect of TLR2 signaling on MDSCs remained controversial. These reports indicated that we still knew little about the mechanisms whereby TLR2 agonists worked on MDSCs in different biological contexts, especially the complicated chronic inflammation environment. Further investigations were needed to clarify this discrepancy.
We have previously demonstrated that heat-killed Mycobacterium bovis BCG-induced pathology-free chronic inflammation triggered the expansion of suppressive CD11b+ Gr-1+ Ly6Gneg Ly6Chigh monocyte-like MDSCs (M-MDSCs) to maintain the internal homeostatic conditions [21]. Data from this study demonstrated that pathology-free chronic inflammation expanded CD11b+ Gr-1+ Ly6Gneg Ly6Chigh M-MDSCs expressing TLR2. Activation of TLR2 signaling enhanced immunosuppression of M-MDSCs by upregulated inducible nitric oxide synthase (iNOS)/nitric oxide (NO) activity partly through signal transducer and activator of transcription 3 (STAT3) activation. These observations confirmed the impact of TLR2 signaling on MDSCs and helped optimize the role of TLR agonists in chronic inflammation by modulating MDSC activity to benefit patients suffering from chronic inflammatory disease.
Material and methods
Mice
C57BL/6 mice were from the Lab Animal Center of Sun Yat-sen University (Guangzhou, China). All mice were maintained under specific pathogen-free conditions in the Lab Animal Center of Sun Yat-sen University. All animal experiments in this study were approved by the Medical Ethics Board and the Biosafety Management Committee of Sun Yat-sen University.
In vivo model system for chronic inflammation
Chronic inflammation was induced by three subcutaneous injection (50 µg per animal/dose) at 1week intervals by heat-killed Mycobacterium bovis BCG (231141; Difco Laboratories). The first two injections were administered as a mixture of heat-killed Mycobacterium bovis BCG and IFA (Sigma) at a 1 : 1 ratio, and the third heat-killed Mycobacterium bovis BCG injection was with phosphate-buffered saline (PBS) only. Spleen cells were collected 2 days after the last injection (day +2).
Suppression assays
T cells were isolated from the spleens of uninfected mice, labeled with carboxyfluorescein succinimidyl ester (CFSE; 1 µM, Life Technologies) for 10 min at room temperature, washed with PBS containing 10% fetal calf serum (FCS), and cultured in 96-well plates (1 × 105 cells/ml) covered with immobilized anti-CD3 mAbs (1 µg/ml) and anti-CD28 mAbs (1 µg/ml). Suppressive MDSCs were enriched for CD11b+ Gr-1+ Ly6Gneg Ly6Chigh populations. Suppressor cells were added to the cultures at a 1 : 1 ratio. Three days later, cells were harvested and stained with PE-anti-CD4, and proliferation of CD4+ T cells was assessed by flow cytometry using CFSE dilution assay. In indicated studies, FACS-sorted suppressor cells were either treated with Pam2CSK4 (1 µM), Stattic (10 µmol/ml) or vehicle (2% carboxymethylcellulose) for 24 h or 72 h. In inhibition experiments, T cells and CD11b+ Gr-1+ Ly6Gneg Ly6Chigh MDSCs were cocultured in the presence of iNOS inhibitors: L-NMMA (10 mM) (Cayman Chemical).
Flow cytometry and cell sorting
MDSCs from the spleen of infected mice were stained for Gr-1, CD11b, Ly6G, Ly6C and TLR2 according to the manufacturer’s instructions. Subsets of MDSCs that were CD11b+ Gr-1+ Ly6Gneg Ly6Chigh or CD11b+ Gr-1+ Ly6Ghigh Ly6Clow cells were purified from spleens of infected mice by a FACS Aria cell sorter (BD Biosciences) using anti-mouse antibodies against Gr-1, CD11b, Ly6G, Ly6C (BD Pharmingen). Sorted cells were reanalyzed on the BD LSRII and the purity of MDSC subsets was shown to be more than 85%. CD11b+ Gr-1+ Ly6Gneg Ly6Chigh MDSCs were co-cultured at 37oC in a CO2 incubator with T cells. Surface staining was performed with Alexa700-labeled anti-CD4 (clone RM 4-5, eBioscience), Alexa 405-labeled anti-CD44 (clone IM7, eBioscience), and PerCP-labeled anti-CD69 (clone H1.2F3, eBioscience). For intracellular cytokine staining, cells were then incubated for 4 h at 37oC in a CO2 incubator with 10 µg/ml brefeldin (eBioscience). Cells were stained with PE-Cy7 labeled-anti-IFN-γ (clone XMG1.2, eBioscience) or PE-Cy7 labeled-anti-iNOS (Clone: CXNFT, eBioscience) after fixation and permeabilization. Respective isotype controls were used to determine non-specific binding. For STAT3 phosphorylation, CD11b+ Gr-1+ Ly6Gneg Ly6Chigh MDSCs were treated with Pam2CSK4 for 24 h and fixed with 2% paraformaldehyde for 10 min at room temperature and permeabilized with 100% ice cold methanol for 30 min at 4oC. Cells were washed and stained with Alexa Fluor 488 labeled anti-STAT3 (pY705) (Clone:4/ P-STAT3, BD Bioscience) for 30 min. Cells were measured on a BD LSRFortessa Flow cytometer and analyzed using FlowJo software (Tree Star).
Nitric oxide production
Nitric oxide (NO) content in plasma was measured following the manufacturer’s protocol (Biovision, Milpitas, CA). An equal volume of supernatant (50 µl) was mixed with Greiss reagent A and B and incubated for 10 min at room temperature in the dark. The absorbance at 550 nm was measured using a microplate plate reader (Bio-Rad, Hercules, CA). Nitrite concentrations were determined by comparing the absorbance values for the test samples to a standard curve generated by serial dilution of 0.25 mM sodium nitrite.
Arginase activity assays
The activity of arginase was measured in cell lysates from isolated MDSCs. Briefly, cells were lysed with 0.1% Triton X-100 for 30 min, followed by the addition of 25 mM Tris-HCl. The enzyme was activated by heating for 10 min at 56oC. Arginine hydrolysis was performed by incubating the lysate with 0.5 M L-arginine and 10 mM MnCl2 at 37oC for 120 min. After the addition of a-isonitrosopropiophenone (dissolved in 100% ethanol), heating at 95oC for 30 min, the urea concentration was measured at 540 nm with a microplate reader (Bio-Rad). Urea contents were calculated based on a serially diluted urea standard curve.
Messenger RNA (mRNA) preparation and RT-PCR
Total RNA was isolated using the RNeasy kit (Qiagen) according to the manufacturer’s instructions. First-strand cDNA synthesis was performed using the Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). The cDNA was quantitated in duplicate using SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) as reported. The sequences of primers for NOS2 were as following: forward: 5'-CTTTGCCACGGACGAGAC-3', reverse: 5'-TCATTGTACTCTGAGGGCTGA-3'. Values were presented as the difference in cycling threshold (Ct) values normalized to those of mRNA encoding β-actin.
Results
Chronic inflammation expanded M-MDSCs expressing TLR2
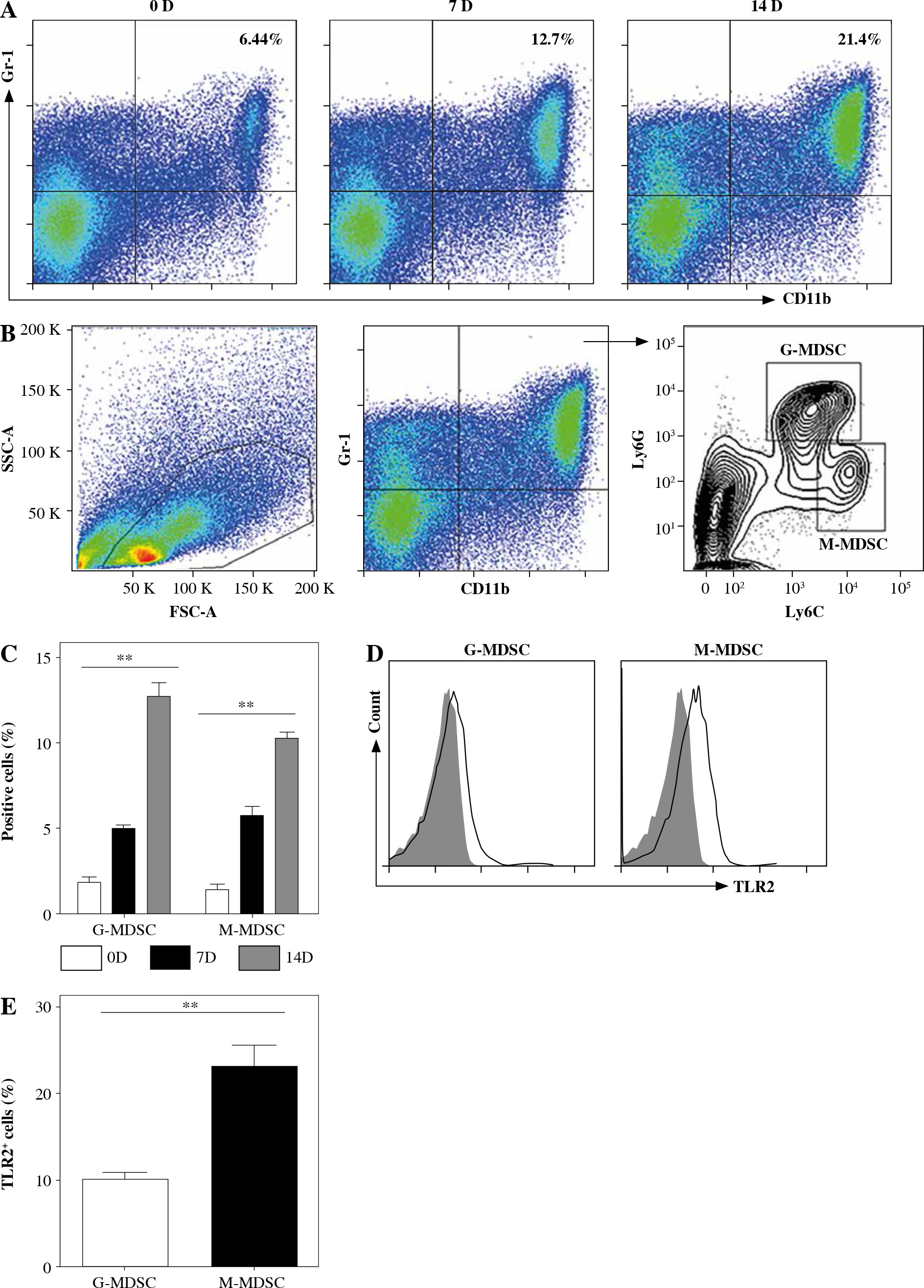
Naive C57BL/6 mice received three subcutaneous injections of heat-killed Mycobacterium bovis BCG. Spleens were harvested for phenotypic analysis of CD11b, Gr-1, Ly6G and Ly6C on the indicated day. As shown in Figure 1A-C, exposure to heat-killed Mycobacterium bovis BCG drove a significant increase of CD11b+ Gr1+ cells at 14 days after immunization. Based on the expression levels of Ly6G and Ly6C, MDSCs are commonly divided into CD11b+ Gr-1+ Ly6Gneg Ly6Chigh monocyte-like MDSCs (M-MDSCs) or CD11b+ Gr-1+ Ly6Ghigh Ly6Clow granulocytic MDSCs (G-MDSCs) respectively. The expression levels of TLR2 were compared between M-MDSCs and G-MDSCs by flow cytometry. Though both subsets of MDSCs expressed TLR2, a significantly higher level of TLR2 expression was observed in M-MDSCs than in G-MDSCs (Fig. 1D, E).
Fig. 1
Chronic inflammation expanded M-MDSCs expressing TLR2. Mice were subcutaneously injected with heat-killed BCG bacteria (50 µg per animal/dose) at 1-week intervals three times. A) Percentages of CD11b+ Gr-1+ myeloid cells in spleen are presented at 0, 7 and 14 days after immunization. B) Flow cytometry analysis show the gating strategy of CD11b+ Gr-1+ Ly6Gneg Ly6Chigh monocytic MDSCs (M-MDSC) and CD11b+ Gr-1+ Ly6Ghigh Ly6Clow granulocytic MDSCs (G-MDSC) after immunization. C) Frequencies of M-MDSC and G-MDSC in spleen at 0, 7 and 14 days after immunization. D) Expression of TLR2 by M-MDSC and G-MDSC at 14 days after immunization. The open histogram represents staining with anti-TLR2 antibody and the shaded histogram represents staining with isotype control antibody. E) Data for percentages of TLR2 in M-MDSC and G-MDSC. Data were obtained from three independent experiments (n = 5 per group). **p < 0.01. Values in (A) were compared using two-way ANOVA with Bonferroni’s test, and values in (E) were compared using t-test
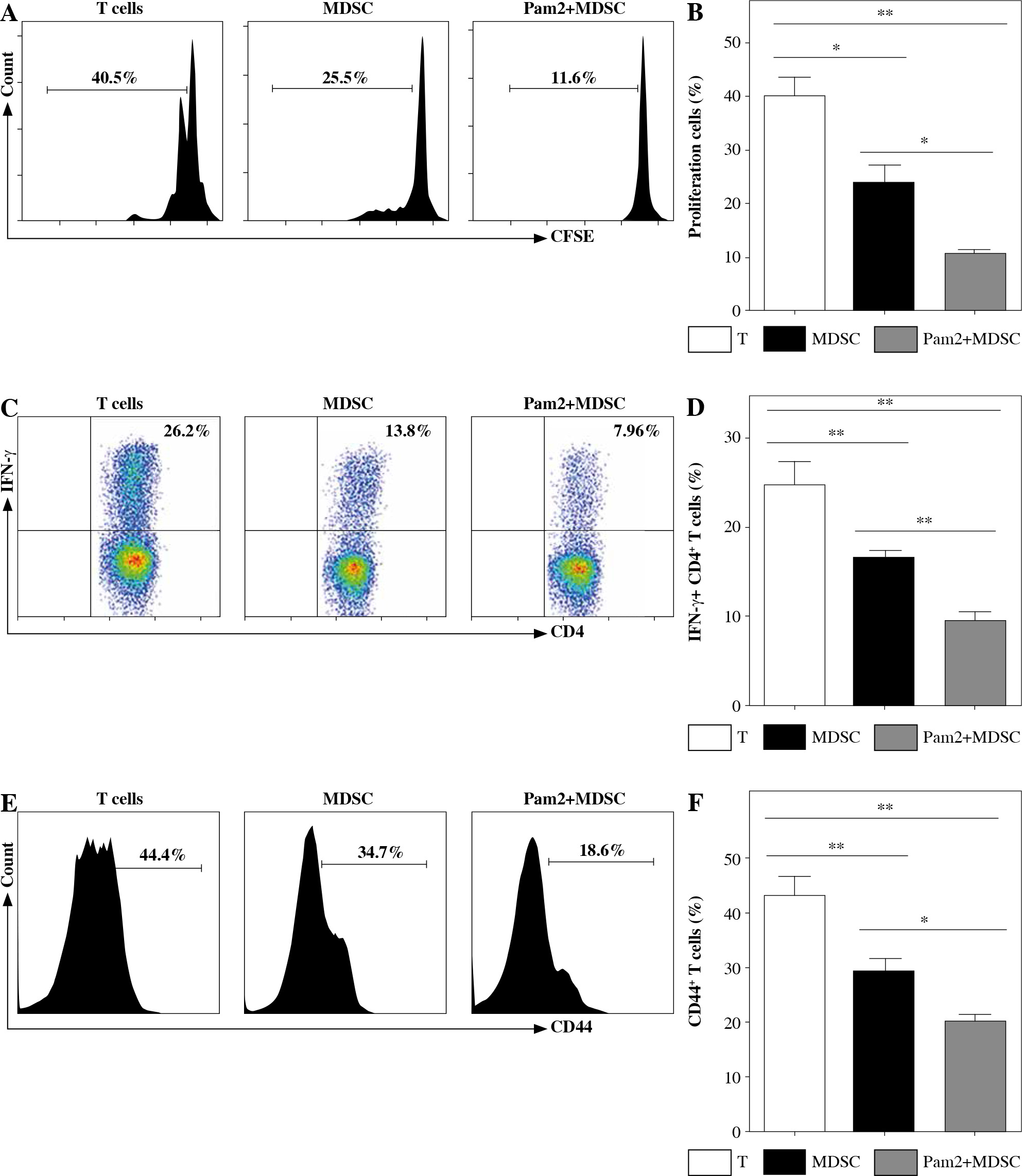
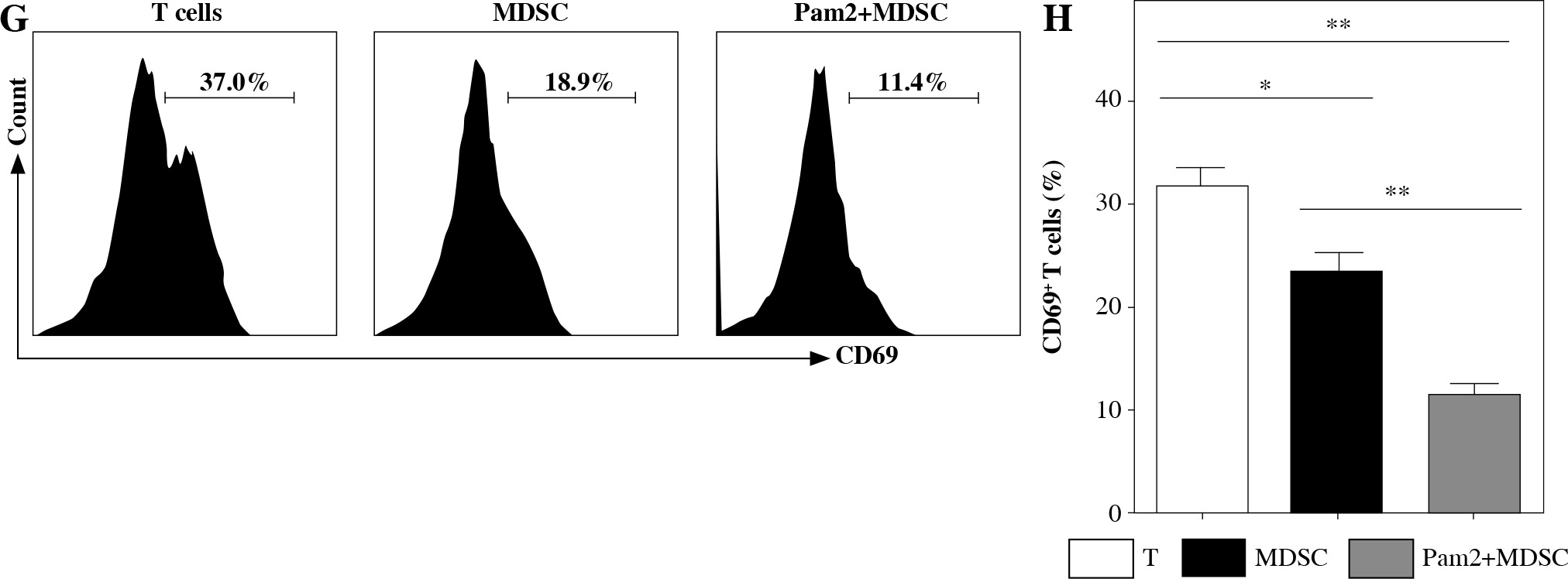
Pam2CSK4 enhanced immunosuppressive activity of M-MDSCs in vitro
To further explore the role of TLR2 in M-MDSCs, we investigated the effect of Pam2CSK4 on the immunosuppressive activity of M-MDSCs. CD11b+ Gr-1+ Ly6Gneg Ly6Chigh monocyte-like MDSCs (M-MDSCs) were enriched using FACS sorting from inflamed mouse. T cells were harvested from spleen of healthy mice, stimulated with anti-CD3/CD28 antibodies, and co-cultured with M-MDSCs at the ratio of 1 : 1 for 3 days with or without Pam2CSK4. The proliferation of CD4+ T lymphocytes was assessed using CFSE dilution assay. T cells were then evaluated for activation markers CD69 and CD44 and cytokine interferon (IFN)-γ production by flow cytometry. The results showed that M-MDSCs efficiently suppressed T cell proliferation and treatment of M-MDSCs with Pam2CSK4 resulted in significantly fewer proliferating CD4+ T cells (Fig. 2A). In addition, Pam2CSK4 treated M-MDSCs more potently impeded IFN-g production in T cells (Fig. 2B). Moreover, M-MDSCs inhibited T cell activation to a lesser extent when compared with their Pam2CSK4 treated counterparts (Fig. 2C, D). Taken together, these data demonstrated that Pam2CSK4 augmented the suppressive activity of M-MDSCs through TLR2 signaling.
Fig. 2
Pam2CSK4 enhanced immunosuppressive activity of M-MDSCs in vitro. CD11b+ Gr-1+ Ly6Gneg Ly6Chigh M-MDSCs (presented as MDSC) were isolated from spleen and enriched using FACS sorting. Purified normal CD4+ T cells were labeled with CFSE and co-incubated with M-MDSC at a ratio of 1 : 1 in the presence of Pam2CSK4 (1 µM) for 72 h. Proliferation, activation and IFN-g production of CFSE-labeled T cells was analyzed. A) Histograms gated on dividing CFSE-labeled CD4+ T. B) Numbers indicate the percentages of proliferation of CD4+ T. C) Representative dot plots show production of IFN-g gated on CFSE-labeled T cells. D) Numbers indicate the percentage of IFN-g+ T cells. E) Representative CD44 and CD69 histograms were gated on CFSE-labeled T cells. F) Numbers indicate the percentage of activated T cells. Data are representative of three independent experiments. *p < 0.05, **p < 0.01. Values in (B, D, F, and H) were compared using one-way ANOVA with Bonferroni’s test G) Representative CD44 and CD69 histograms are gated on CFSE-labeled T cells. H) Numbers indicate the percentage of activated T cells. Data are representative of three independent experiments. *p < 0.05, **p < 0.01. Values in (B, D, F, and H) were compared using one-way ANOVA with Bonferroni’s test
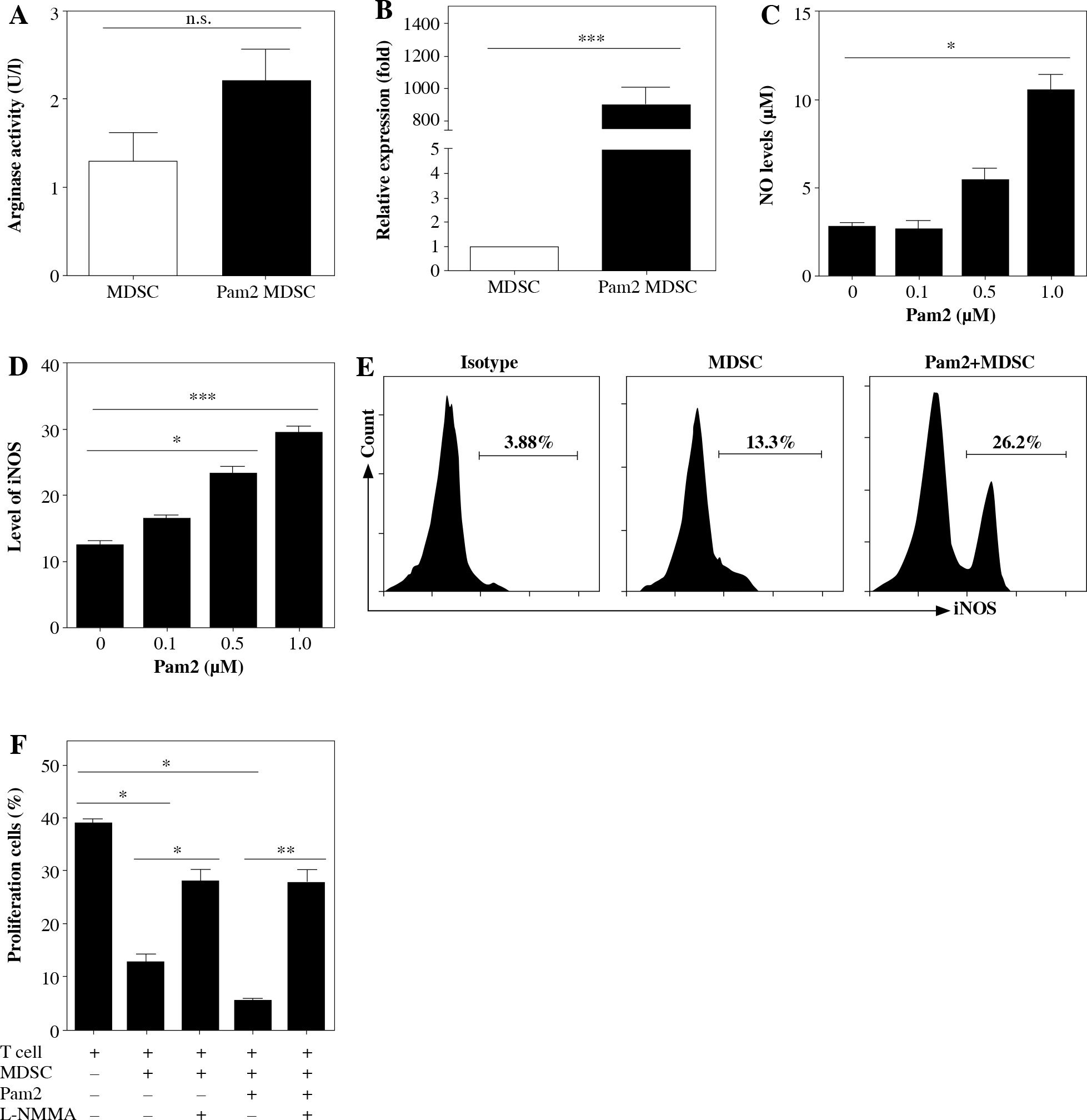
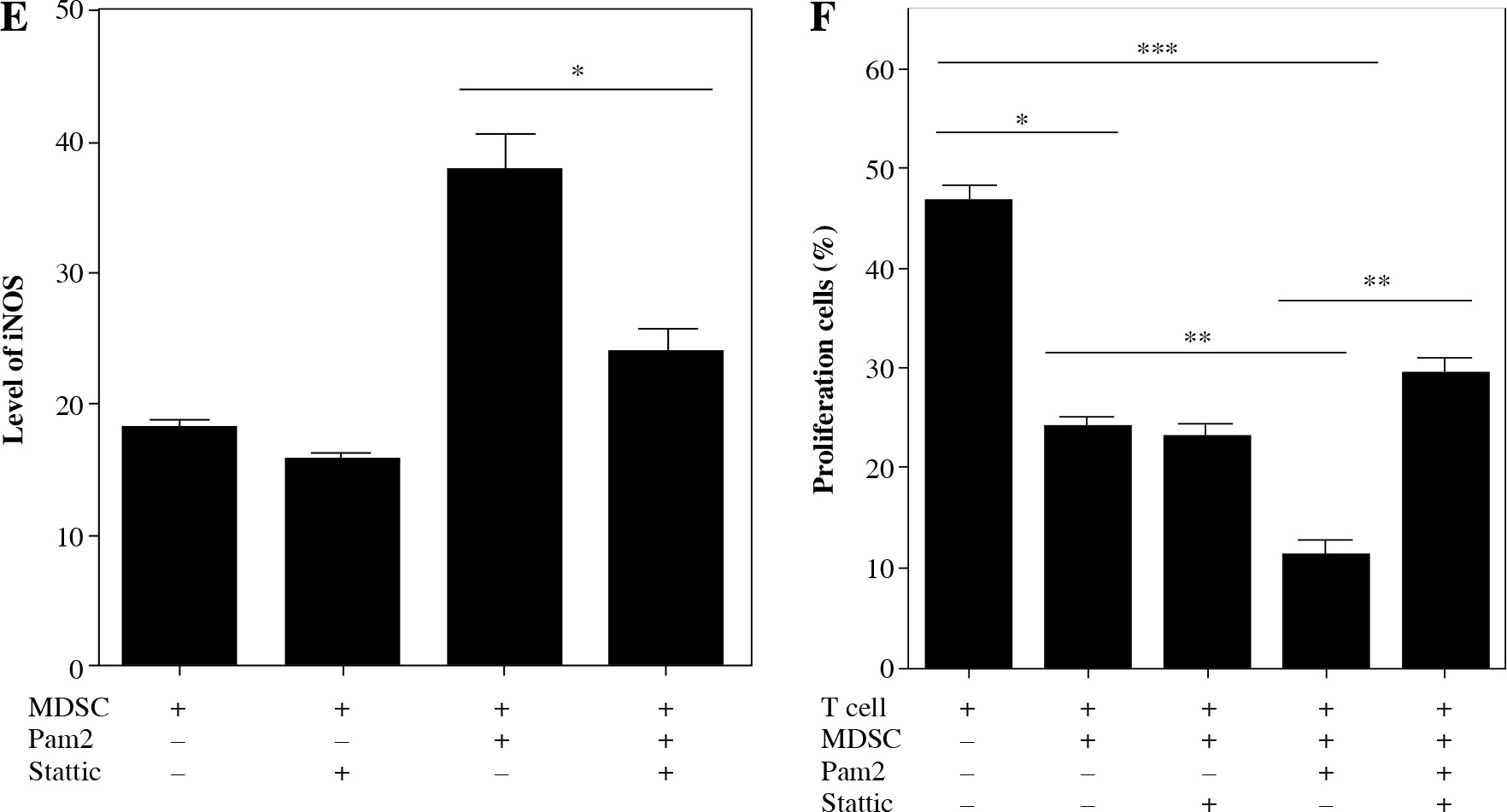
iNOS activity was essential for Pam2CSK4-enhanced suppressive activity in M-MDSCs
Next, we investigated the mechanism underlying the enhanced suppressive function of Pam2CSK4 treated M-MDSCs. Arginase and NO were critical molecules responsible for the suppressive activity of MDSCs. Thus, we examined the effects of Pam2CSK4 on the activities of arginase or iNOS in MDSCs. However, there was a non-significant trend towards an increase in ARG-1 activitity in Pam2CSK4 treated M-MDSCs (Fig. 3A). These data indicated that arginase activity was a minor contributor to stronger suppressive function of Pam2CSK4 treated M-MDSCs. Meanwhile, the expression level of NOS2 (Fig. 3B), the production of NO (Fig. 3C) and the activity of iNOS (Fig. 3D, E) of M-MDSCs were all significantly enhanced by Pam2CSK4. Moreover, the increase suppressive activity mediated by Pam2CSK4 was largely abrogated by the iNOS inhibitor L-NMMA (Fig. 3F). These data suggested that iNOS/NO is critical for the enhanced suppressive function of M-MDSCs with Pam2CSK4.
Fig. 3
iNOS was essential for Pam2CSK4-enhanced suppressive activity in M-MDSCs. CD11b+ Gr-1+ Ly6Gneg Ly6Chigh M-MDSCs (presented as MDSC) were enriched using FACS sorting and treated with or without Pam2CSK4 (1 µM) for 24 h ex vivo. A) Arginase activity of MDSCs. B) Quantitative RT-PCR analysis of NOS2 gene expression by MDSCs. C) Level of NO in cultured supernatant in MDSCs treated with Pam2CSK4 for 24 h at indicated concentration 0.1 µM, 0.5 µM and 1.0 µM ex vivo. D) Data of activity of iNOS of MDSCs treated with Pam2CSK4 for 24 h at indicated concentration 0.1 µM, 0.5 µM and 1.0 µM ex vivo. E) Representative histograms of activity of iNOS in MDSCs with Pam2CSK4 (1 µM) treatment for 24 h. F) CD4+ T cells were stimulated with anti-CD3/CD28, co-cultured with MDSCs at a 1 : 1 ratio in the presence of Pam2CSK4 (1 µM) for 72 h and evaluated for T cell proliferation by CFSE. Data show the effect of iNOS inhibitor L-NMMA (10 mM) on MDSC function in the presence of Pam2CSK4. Data for the percentages of proliferative CD4+ T cells were obtained from three independent experiments (n = 5 per group). T cells alone were used as a positive control. *p < 0.05, **p < 0.01, ***p < 0.005, n.s. – non-significant. Values were compared using t-test
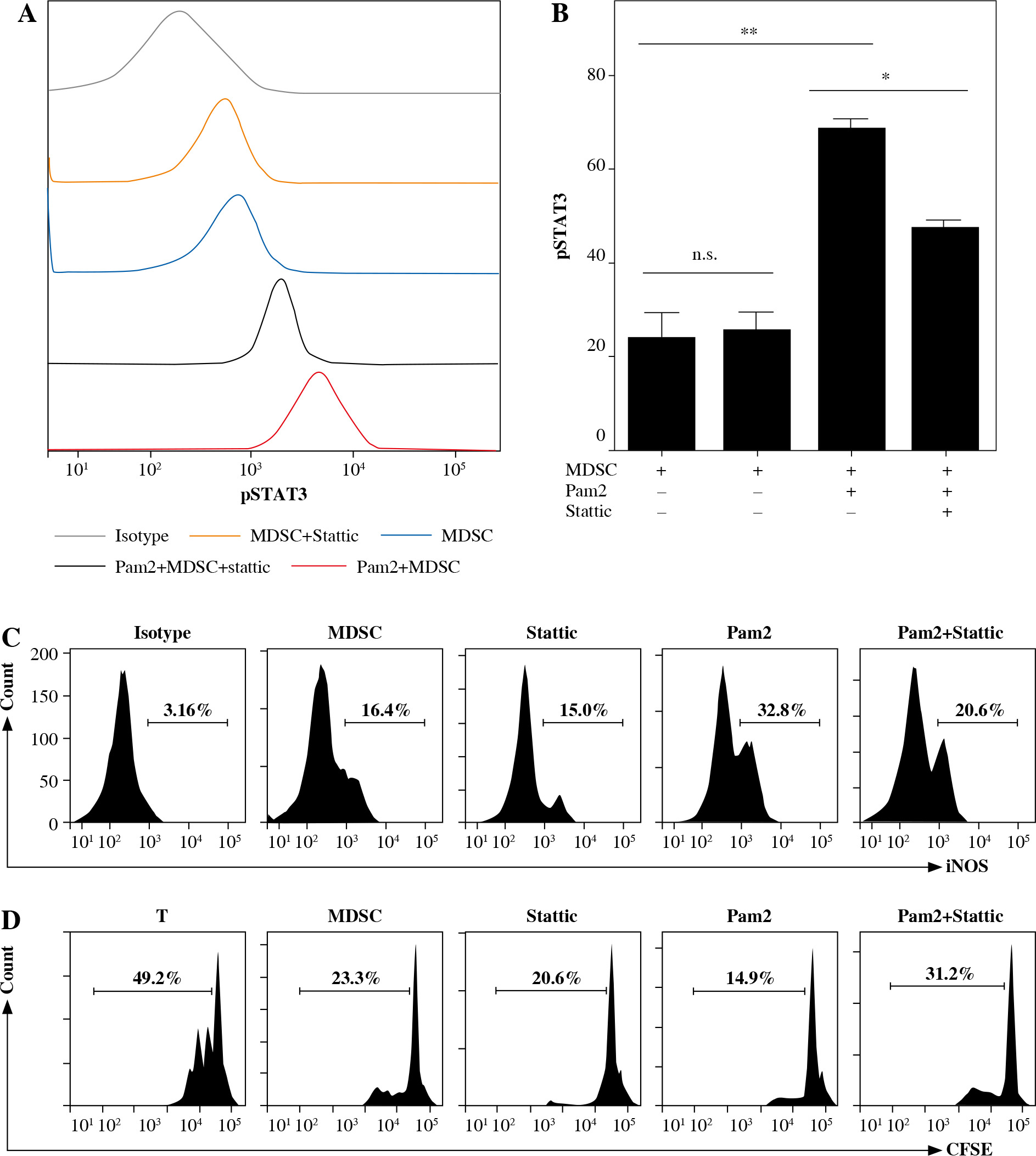
STAT3 mediated iNOS activity in Pam2CSK4-treated M-MDSCs
Given the functional significance of the STAT3 signal in MDSCs, we analyzed intracellular phosphorylated STAT3 in M-MDSCs with or without Pam2CSK4 treatment in the presence or absence of Stattic, a specific small molecule inhibitor of pSTAT3. Interestingly, Pam2CSK4 treated M-MDSCs had significantly higher expression levels of pSTAT3 compared with counterparts without Pam2CSK4 (Fig. 4A, B). The results showed that Stattic was capable of inhibiting pSTAT3 in Pam2CSK4 treated M-MDSCs but had no impact on M-MDSCs without Pam2CSK4. Stattic decreased the activity of iNOS in Pam2CSK4 treated M-MDSCs (Fig. 4C, E), and partly restored T cell proliferation (Fig. 4D, F). These results suggested that STAT3 activation was required for the higher iNOS activity, and contributed to stronger suppressive function of Pam2CSK4 treated M-MDSCs.
Fig. 4
STAT3 mediated iNOS activity in Pam2CSK4-treated M-MDSCs. CD11b+ Gr-1+ Ly6Gneg Ly6Chigh M-MDSCs (presented as MDSC) were enriched using FACS sorting and treated with Stattic or vehicle in the presence or absence of Pam2CSK4 for 24 h ex vivo. A) Expression of phosphorylated STAT3 in M-MDSCs stimulated for 24 h with Stattic or vehicle in the presence or absence of Pam2CSK4. B) Data are representative of three independent experiments in A. C) Intracellular iNOS expression induced by overnight stimulation with Stattic or vehicle in the presence or absence of Pam2CSK4. D) CD4+ T cells were stimulated with anti-CD3/CD28 and co-cultured with M-MDSCs at a 1 : 1 ratio with treatments as indicated. T cell proliferation was measured by CFSE dilution assay. Histograms comparing the proliferation of CD4+ T cells. *p < 0.05, **p < 0.01, ***p < 0.005. Values were compared using t-test. E) Data are representative of three independent experiments in C. F) Data for the percentages of proliferative CD4+ T cells were obtained from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.005. Values were compared using t-test.
Discussion
Chronic inflammation originating in a response to any stimulus might induce immunosuppression to maintain the internal homeostatic conditions. MDSCs are known to be one of the main mediators orchestrating the suppressive environment [22]. Recent reports have suggested that the survival, differentiation, and suppressive activity of MDSCs were influenced by TLR signaling [23]. Due to the plasticity and diversity of MDSCs, TLR-induced MDSCs could be beneficial or detrimental to the outcome of disease depending on the location, nature, and the stage of the ongoing chronic inflammatory process. Our study demonstrated that heat-killed Mycobacterium bovis BCG-induced chronic sterile inflammation expanded TLR2+CD11b+ Gr-1+ Ly6Gneg Ly6Chigh M-MDSCs. TLR2 stimulation of M-MDSCs by treatment with Pam2CSK4 activated STAT3 and induced iNOS/NO, thus contributing to the enhanced immunosuppressive activity of M-MDSCs.
Research on cancer, infection and autoimmunity demonstrated that TLR2 signaling promoted the expansion, accumulation, differentiation and activation of MDSCs via different pathways [3, 13-17]. Recent studies demonstrated that tumor-derived soluble factors HSP72 promoted tumor growth by inducing expansion and recruitment of MDSCs through activation of STAT3 in a TLR2/MyD88-dependent manner, which was triggered by autocrine production of interleukin (IL)-6 [24]. Moreover, serum amyloid A3 induced TLR2 signaling and enhanced the suppressive activity of M-MDSCs through STAT3 activation, by autocrine secretion of tumor necrosis factor α (TNF-α) [15]. Therefore, TLR2 activation by specific agonists increases the potential of MDSCs to suppress anti- tumor immune responses. Similarly, HCV induced MDSC-like suppressive monocytes through the TLR2/PI3K/AKT/STAT3 signaling pathway, consequently inducing CD4+ Foxp3+ regulatory T cells and inhibiting autologous CD4+ T cell activation [25]. The TLR2 pathway could also be specifically involved in the expansion of G-MDSCs in mice with adjuvant-induced arthritis [3]. Thus, the above cumulative research indicated that TLR2 influenced MDSCs by different pathways depending on the environment to which they were recruited.
In the present study, higher expression of TLR2 was observed in M-MDSCs than G-MDSCs in heat-killed BCG inducing sterile chronic inflammation. This finding was in line with the observation that M-MDSCs exhibited higher TLR2 expression than other immune cells in the tumor microenvironment [4, 18]. However, further investigations were needed to illuminate the mechanism underlying this phenomenon. Pam2CSK4 treated M-MDSCs exhibited enhanced STAT3 phosphorylation. Notably, treatment with Stattic, a specific small-molecule inhibitor of STAT3, significantly abrogated the suppressive activity of M-MDSCs. It has been shown that the signaling pathways responsible for the induction of iNOS and NO secretion in MDSCs triggered by TLRs were alike in proceeding via MyD88 and nuclear factor kappa B (NF-κB) [26, 27]. In this regard, our data suggested that STAT3 activation might partly contribute to the suppressive function of TLR2+ M-MDSCs during chronic inflammation. According to previous reports, the combination of LPS and GM-CSF induced differentiation of lin-progenitor cells into MDSCs [28]. Moreover, STAT3 was reported to differentially regulate early-and late-phase TLR4-mediated inflammatory responses [29]. Therefore, a possible mechanism through which STAT3 was activated in Pam2CSK4 treated M-MDSCs involved the co-operation between TLR/MyD88 and JAK/STAT pathways in MDSC development. Indeed, following constitutively TLR-dependent stimulation, NF-κB was activated and ensured transcription of target inflammatory genes, such as IL-6 and TNF-α, which regulated the generation and function of MDSCs, primarily via the JAK/STAT3 signaling pathway [30]. Therefore, it was not surprising that prolonged and persistent exposure to heat-killed BCG triggered STAT3 activation in M-MDSCs through the TLR/MyD88 pathway. Taken together, these results confirmed the role of the TLR2 driven program in MDSCs during chronic inflammation.
iNOS-derived NO has been associated with the pathogenesis and progression of several diseases. iNOS has been related to pathological processes associated with immuno- activation and inflammation [31]. The bulk of evidence suggested that the iNOS/NO pathway could be considered as a parameter that promoted chronic inflammation and aging. Different mechanisms might account for the production of NO [32]. Current knowledge on the role of STAT3 concerning regulation of iNOS and NO is limited. STAT3 was traditionally shown to regulate the effect of ARG1 and was not required for iNOS activity, which was driven by STAT1 and NF-κB signaling activated in M-MDSCs [33]. In the present study, we found that STAT3 was also responsible for iNOS/NO associated immunosuppression of M-MDSCs through Pam2CSK4/TLR2 activation. In our opinion, these data were not in contrast but reflected the multifaceted involvement of STAT3 in MDSCs function, which might be context-dependent. A hint toward this hypothesis was conferred by the observation that p-STAT3 was able to bind iNOS promoter sites to enhance the production of NO on astrocytes [34]. Indeed, it has been reported that STAT3 modulated iNOS activity and NO secretion to a different extent in dendritic cells [35], cancer cells [36, 37] and Raw264.7 cells [38]. Consistent with our results, it was reported that both IL-6 and IL-10 critically contributed to ascitic fluid driven expansion of CD14+ HLA-DR–/low M-MDSCs in a STAT3 dependent manner, which executed the immunosuppressive mechanism partly through iNOS [39].
It should be pointed out that the present study has several limitations, including the single animal model, insufficient sample size, and the lack of patient investigations. The majority of experimental studies on the role of chronic inflammation have been conducted using models of carcinoma [13, 24] or infections [40], whereas we used the model of heat-killed BCG induced sterile inflammation [41]. We suspect the perceived discrepancy that STAT3 regulated the effect of iNOS rather than ARG-1 in M-MDSCs could be ascribed to the mycobacterial strain-specific differences and the surrounding inflammatory milieu. Indeed, a complete understanding of the TLR2 related pathway modulating MDSCs induction and function would assist in the development of its therapeutic application. Therefore, more sophisticated investigation with in vivo models using animals deficient in TLR2 or STAT3 was required to provide more reliable evidence to support the conclusion that TLR2 regulated M-MDSCs via the STAT3/iNOS pathway. Moreover, since the molecular basis and the effect of biochemical variation in the regulation of TLR signaling on the expansion of MDSCs might be different between mouse and human, a large cohort of patients with various chronic disorders induced by sterile inflammation is also needed to validate the clear molecular mechanisms.
In summary, heat-kill BCG triggered chronic inflammation induced suppressive M-MDSCs expressing TLR2, which executed the immunosuppressive mechanism by iNOS/NO partly in a STAT3 dependent manner. Our data clearly demonstrated that TLR2 activation of MDSCs played an important role for the maintenance of immune system homeostasis by avoiding excessive immune stimulation in chronic inflammation. Further studies are needed to verify whether the TLR2/STAT3/iNOS axis in MDSCs might represent a potential therapeutic target in chronic inflammatory disease.