Introduction
Shijiazhuang, the provincial capital of Hebei province, suffers from serious air pollution, especially with the fine particulate PM2.5, and it is also plagued by cardiovascular diseases. The two phenomena are likely causally related, creating widespread public concern [1, 2]. Atherosclerosis, the principle pathology of cardiovascular disease, is thought to represent a chronic inflammatory disease. Circulating leukocytes and platelets participate in inflammatory responses that drive atherogenic expansion [3]. Until recently, air pollutants had been regarded as risk factors for atherosclerosis, particularly the air contaminant PM2.5. It is thought that PM2.5 evades respiratory system barriers and enters the circulation directly, where it alters the functions of leukocytes, induces oxidative stress, and disrupts innate immunity mechanisms [4, 5]. Nevertheless, the mechanisms leading to atherosclerosis have not been fully elucidated.
Circulating leukocytes, mainly monocytes and macrophages, are components of the innate immunity system responsible for maintaining physiologic equilibrium and monitoring extraneous materials by inducing inflammatory responses. This mechanism evolved to clear foreign substances [6, 7]. Under pathological conditions, persistent or uncontrolled inflammation may cause host organ damage and atherosclerosis, leading to myocardial infarction and stroke. The initial phase of atherosclerosis appears to be damage to endothelial barriers at arterial bifurcations under hyperlipidemic conditions [8]. Circulating platelets are recruited immediately and express adhesion factors and receptors, including P-selectin, CD40L, VI and glycoprotein Ib, directing platelet-leukocyte interaction to facilitate leukocyte accumulation and activation, creating a vicious cycle [6, 7, 9]. Circulating monocytes migrate into lesions and subsequently differentiate into macrophages, the major cellular component in plaques. Heterogeneous alterations in macrophage secretory function determine the progression of atherosclerosis. Generally, macrophages can be divided into two sub-populations: classically activated macrophages (M1) release abundant inflammatory cytokines as well as oxidative stress, causing leukocyte recruitment, activation, differentiation and survival. Most prominent among these inflammatory cytokines are tumor necrosis factor α (TNF-α), IL-6, IL-1β, and type I interferon (IFN-α, β); the second sub-population consists of alternatively activated macrophages (M2) that elaborate anti-inflammatory and anti-atherogenic effects [10, 11]. Therefore, the balance between M1 and M2 macrophages is critical for development or inhibition of atherosclerosis. PM2.5 is a newly discovered inducer of atherosclerosis. It presumably promotes oxidative stress and visfatin expression. Its effector molecules are currently under investigation.
Material and methods
Preparation of animals
Eight-week-old male apoE–/– mice were purchased from Cavens Laboratory Animal Co, Ltd (Changzhou, China). All mice were fed a high-fat and cholesterol-rich atherogenic diet (containing 21% fat, 19.5% casein, and 1.25% cholesterol) for 8 weeks at 20–24°C and 45–55% humidity with a 12-h light-dark cycle. The mice were divided randomly into the PM2.5 group (n = 16) and the control group (n = 16). Mouse body weight was 20 ±2 g. In the PM2.5 group, 10 mg of purified power of PM2.5 was suspended in 4.8 ml of carboxy methyl cellulose sodium (CMC) solution for daily dosages, and every mouse received 0.3 ml of solution by gavage once a day for 8 weeks with a high-fat diet. The control group received the same dosage of CMC. All manipulations of animals conformed with the animal ethics committee of Hebei General Hospital as well as the NIH Guide for the Care and Use of Laboratory Animals.
PM2.5 preparation
The particulate matter (PM) was provided by Hebei Provincial Center for Disease Control and Prevention in Shijiazhuang, China. The PM2.5 samples were prepared using the PM2.5 air sampler and were collected on membrane filters for 24 h/day. PM2.5 membrane filters were weighed and dissolved in normal saline and were further sonicated for 30 min to scatter the particles and obtain supernatant fluid according to previous studies [5]. Finally, PM2.5 was frozen by liquid nitrogen, dried, and stored at 4°C in the dark [4].
Measurement of serum glucose, lipids, IL-6 and TNF-α levels
Blood removed from the eye of anesthetized mice was collected in procoagulant to obtain serum. Serum glucose levels were examined using the glucose oxidase method (Beckman). Total cholesterol (TC), triglycerides (TG), low-density lipoprotein-cholesterol (LDL-C), high-density lipoprotein-cholesterol (HDL-C), IL-6 and TNF-α levels were tested by ELISA kits from Neobioscience Diagnostics.
Measurement of whole blood heavy metals
Ten mg/l standard solutions of aluminum, vanadium, iron, manganese, selenium and silver (Agilent Technologies Inc, California, American) were used to formulate various mass concentrations of 0.00, 5.00, 10.00, 20.00, 50.00, 100.00 and 200.00 μg/l to build a standard curve, and then the whole blood levels of appropriate heavy metals were determined by inductively coupled plasma-mass spectrometry (ICP-MS, Thermo Fisher Scientific, Massachusetts, USA) directly.
Oil Red O staining for the descending aorta
To evaluate the lipid deposition in whole arteries, the aortas were dissected from the left subclavian artery to the iliac bifurcation, then opened longitudinally and stained with Oil Red O for 30 min and washed with 75% alcohol. Aortic images were captured with a Sony DXC-960MD (Sony Corporation, Tokyo, Japan), and the lesion size (red positive areas) was quantitatively calculated. Data were analyzed by Image-Pro Plus-6 software (Media Cybernetics, Inc).
Scanning electron microscopy (SEM)
The brachiocephalic arteries (BCA) were dissected and fixed with electron microscope perfusion fixative (2.5% glutaraldehyde in Millonig’s phosphate buffer) for 72 h and then dehydrated by increasing grades of ethyl alcohol and subsequently dried with a LPD-100 critical point dryer (S4800; Hitachi Limited, Inc., Tokyo, Japan) for 5 min at 41°C and 1,200 psi CO2. The specimens were then coated on aluminum stubs with silver glue and observed by SEM [12, 13].
Immunohistochemistry, Masson and Sirius red staining
Aortic roots were sliced into 5 μm sections continuously for pathological evaluation. Masson (SS0236; Xinhua Luyuan Science and Technology Ltd, Beijing, China) and Sirius red (DC0041; Leagene Biotechnology Co., Ltd, Beijing, China) staining was performed according to installation instructions to evaluate plaque areas and collagen separately. Sections for immunohistochemistry were first subjected to microwave retrieval and incubated with corresponding antibodies at 4°C overnight and subsequently labeled with horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin G (IgG) at room temperature for 45 min. Finally, they were visualized by DAB and observed by upright microscopy (Carl Zeiss). Antibodies against α-actin (1 : 150 dilution) and galectin-3/MAC-2 (1 : 200 dilution) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Q-PCR
Total RNA from the abdominal aorta was isolated using TRIzol (Invitrogen), then treated with DNase I at 37°C for 30 min before reverse transcription using a cDNA archive kit (TaKaRa). The cDNA preparations were tested by real-time PCR based on SYBR Green methods, according to the manufacturer’s instructions. The amplification and detection of specific products were performed according to the manufacturer’s protocol with the IQ5 system (Bio-Rad). Associated gene primers are shown in the supplement.
Echocardiography and hemodynamic measurements
Transthoracic echocardiography was performed at the end of our experiment. Briefly, prior to ultrasound imaging, mouse thoraxes were shaved and animals were anesthetized with 1% isoflurane. We used visual sonic vevo770 (VisualSonics, Toronto, Canada). Thirty MHz high-frequency head scans and associated linear signal transducers were used. The following parameters were recorded: heart rate, left ventricular end systolic dimension (LVESd), left ventricular end diastolic dimension (LVEDd), left ventricular ejection fraction (LVEF), fractional shortening (FS), left ventricular end diastolic posterior wall thickness (LVPWd), and left ventricular end diastolic anterior wall thickness (LVAWd). Images were analyzed during end-diastole and during end-systole.
Results
Blood levels of heavy metal content in PM2.5-treated apoE–/– mice and composition of heavy metals of purified PM2.5 powder
PM2.5 particulate matter is derived from the burning and emissions from fossil fuel or industrial production. Most toxic heavy metals float in the air, especially aluminum, iron, chromium and arsenic, the major toxic substances. We found remarkably elevated heavy metal levels in whole blood of the PM2.5 group (Table I), especially aluminum and iron. The accurate heavy metal composition of PM2.5 was also measured (Table II), and the proportions were similar to those of whole blood (Table I). These data indicated that PM2.5 heavy metal particles could enter the circulating system by gavage treatment, suggesting further research pathways.
Table I
Effect of PM2.5 administration on levels of heavy metal contents in whole blood of apoE–/– mice
PM2.5 had no significant effects on serum glucose and lipids, but increased pro-inflammatory factor levels
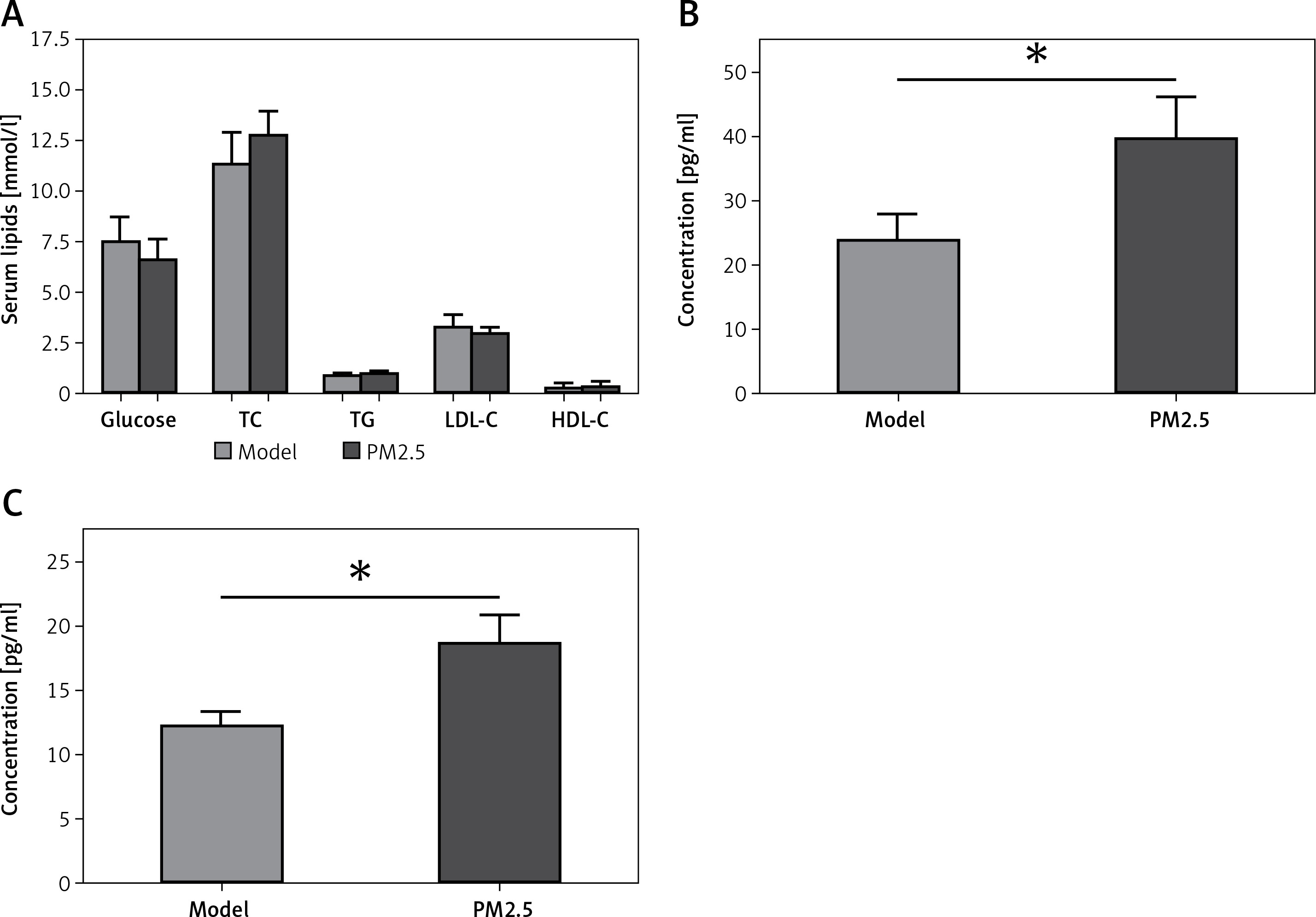
There were no significant differences in levels of serum glucose, TC, TG, LDL-C or HDL-C between the PM2.5 and control groups (Figure 1 A). However, apoE–/– mice treated with PM2.5 showed higher serum levels of pro-inflammatory factors (IL-6 and TNF-α) than did controls (Figure 1 B). Our results suggested that PM2.5 amplified inflammatory responses independently of serum lipid levels.
Figure 1
Treatment of apoE–/– mice with PM2.5 induced increased serum pro-inflammatory factor levels but had no significant effects on serum glucose and lipids. ApoE–/– mice were fed a high-fat, atherogenic diet containing 1.25% cholesterol and 21% fat, and were administered PM2.5 (30 mg/kg/day) for 8 weeks. Serum glucose, TC, TG, LDL-C and HDL-C (A, p = 0.67, 0.6915, 0.9495, 0.5612, 0.7851, respectively), as well as IL-6 and TNF-α (B, C, p = 0.0196, 0.0367) were measured by ELISA
Data represent the mean ± SE (n = 8). *P < 0.05 versus control.
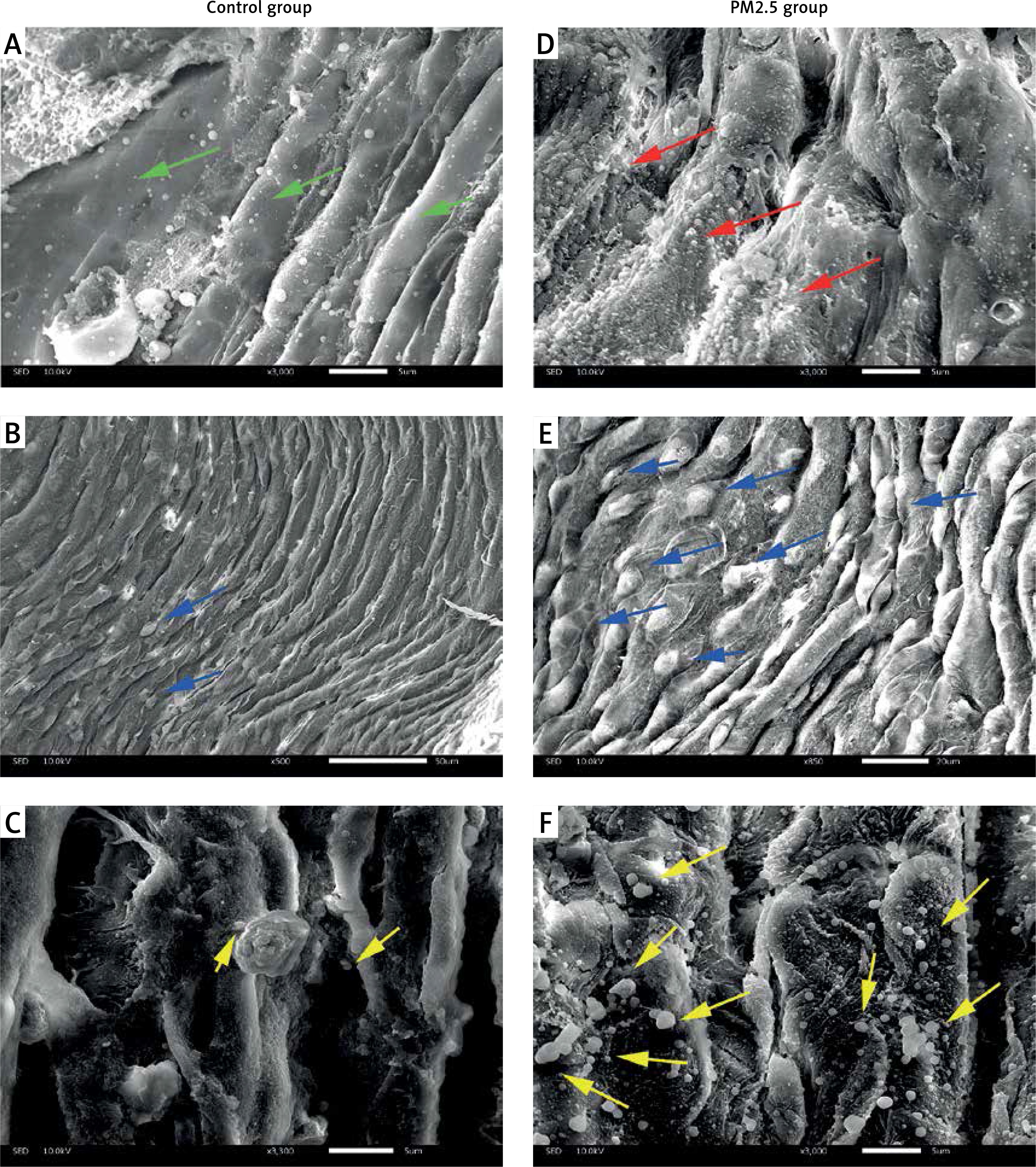
PM2.5 deteriorated atherosclerotic status observed by SEM
Brachiocephalic arteries (BCA) are the most atheroma-prone vessels in apoE–/– mice. Therefore, we chose BCA for SEM assessment of atherosclerotic surfaces. Compared with the smooth epidermis of control mice (green arrows in Figure 2 A), surfaces were “rougher” (indicated by red arrows in Figure 2 D) in the PM2.5 group. In addition, we found more adherent leukocytes in experimental tissues (indicated by blue arrows in Figures 2 B and E) shown as bulging and distended “balls” appearing on the endothelial surface. Most notably, we observed abundant platelets (indicated by yellow arrows, Figures 2 C, F). In conclusion, increased amounts of adherent leukocytes and platelets on the atheroma surface were found after PM2.5 exposure.
Figure 2
PM2.5 administration induced invasion by leukocytes and platelets, and impaired endothelial surfaces in BCA. Scanning electron microscopy was performed to evaluate the status of the surface of atheroma-prone BCA. More smooth endothelial cells were found in the control (A, green arrows) and rough or damaged surfaces were common in the PM2.5 group (D, red arrows); PM2.5 administration resulted in widespread platelet adherence (F, yellow arrows); adhered leukocytes (E, blue arrows) were found on the surface of arteries
PM2.5 amplified atherosclerotic lesions in apoE–/– mice
We further evaluated the effect of PM2.5 on the size of atherosclerotic lesions in apoE–/– mice. Oil Red O stained en face specimens of total descending arteries (initiated from the aortic arch to the bifurcation of the common iliac artery) were used to represent areas occupied by atherosclerotic plaques, measured by the quantitative morphological method. There was a higher percentage of aortic area (lesion area compared to total arch area) in apoE–/– mice treated with PM2.5, compared with controls (Figures 3 A and B). Similarly, we also found increased plaque areas in aortic roots of the PM2.5 group as shown by Masson staining (Figures 4 Aa and Ba). These data suggest that PM2.5 treatment facilitated lesion size expansion in apoE–/– mice.
Figure 3
PM2.5 raised areas of atherosclerotic lesions in apoE–/– mice. The ratios of whole aortic lesion areas (Red-positive areas compared to total vessel areas) were measured by quantitative histology of Oil Red O stained en face samples (A and B, n = 8, p = 0.0276)
Data represent the means ± SE.*P < 0.05 versus control.
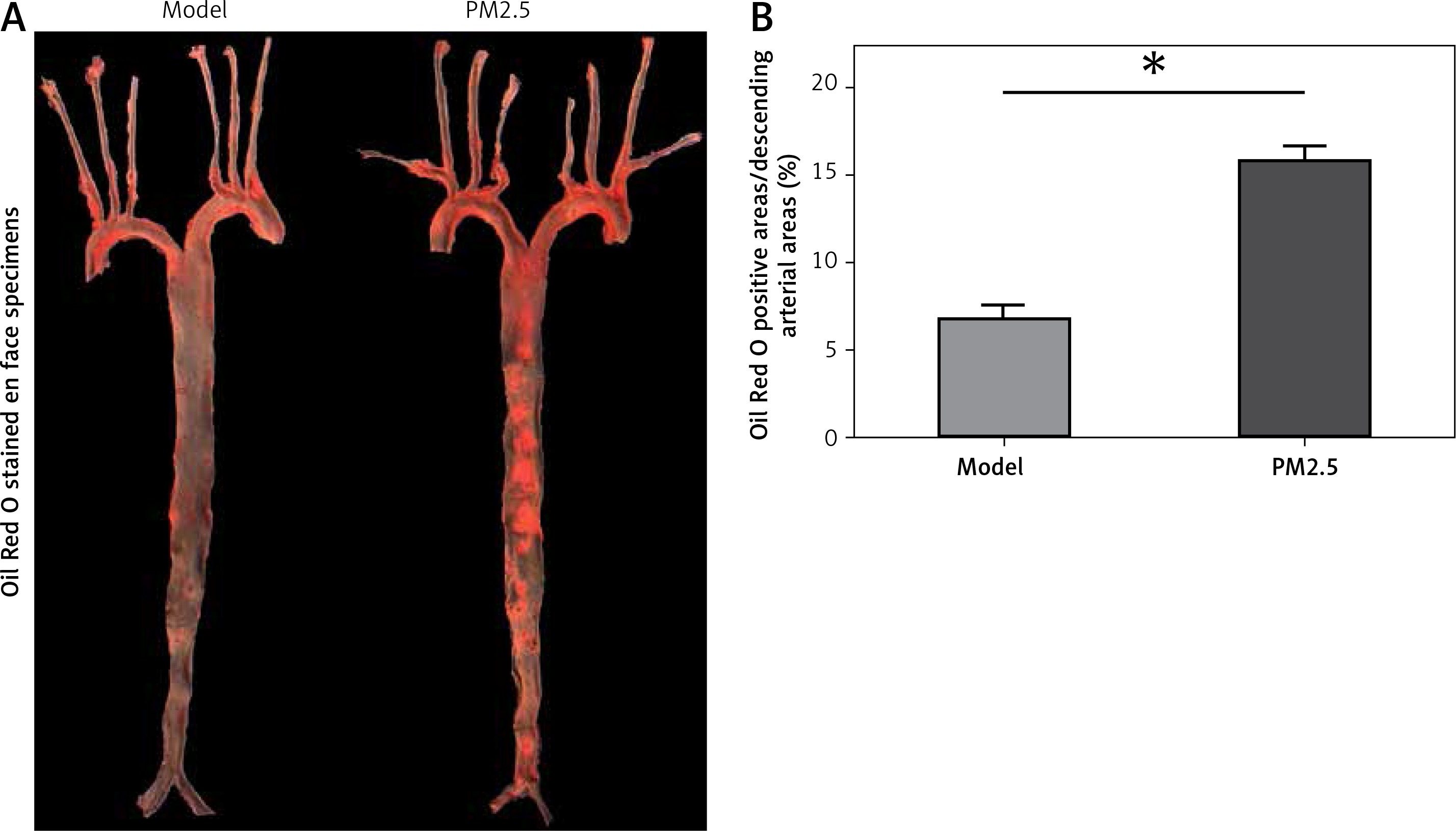
Figure 4
PM2.5 altered lesion areas and cellular composition in aortic roots of apoE-/- mice. Lesion areas in sections from aortic roots were stained with Masson trichrome (Aa, p = 0.0231). The quantities of macrophages (Ab, p = 0.0183), SMC (Ac, p = 0.6355) and content of collagen (Ad, p = 0.025) in the plaques of aortic roots were determined by immunohistochemistry and Sirius Red staining, respectively
Data represent the means ± SE (n = 8). *P < 0.05 versus control.
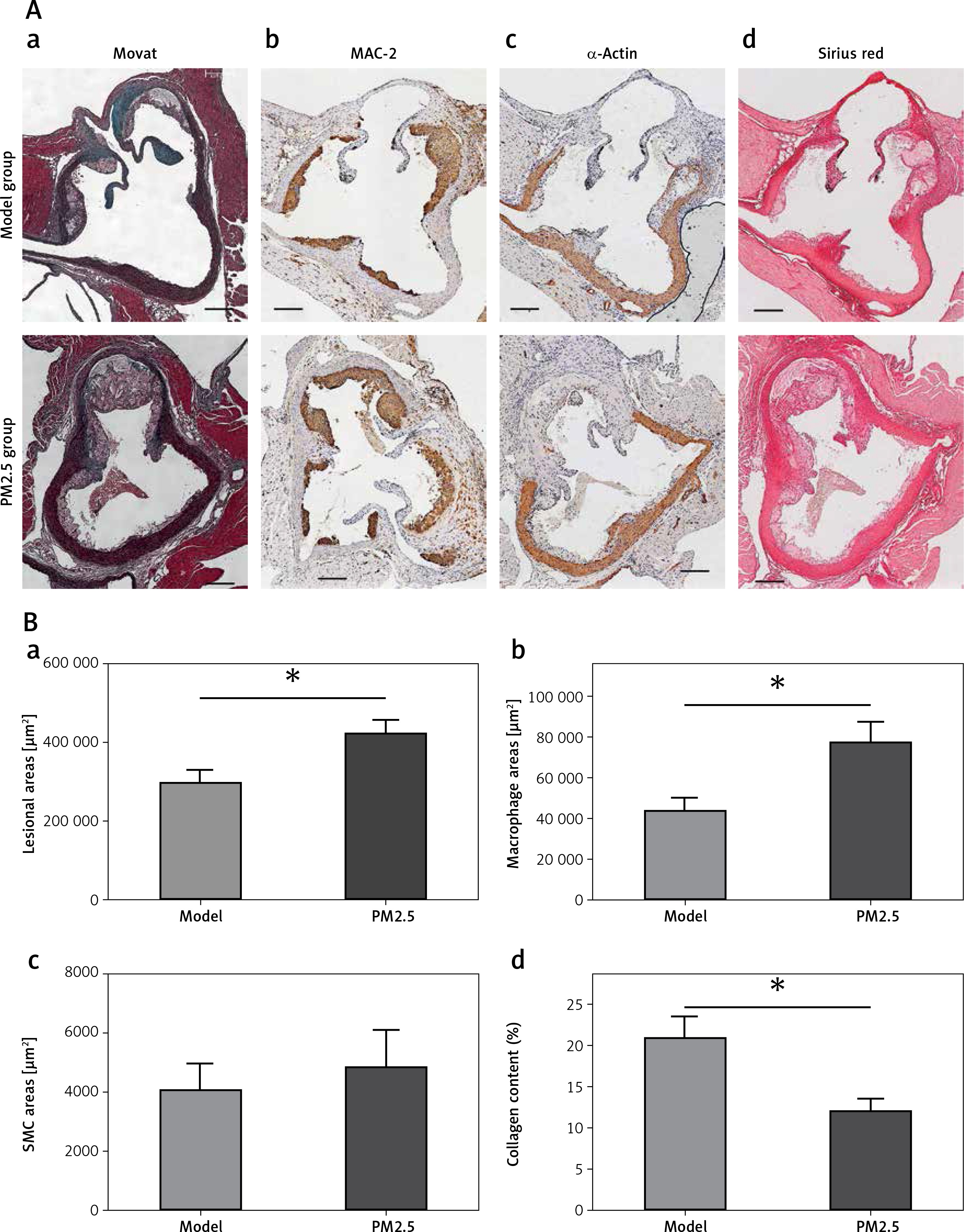
PM2.5 altered lesion cellular contents in apoE–/– mice
Cellular composition and plaque contents determine the stability of atherosclerotic plaques, particularly macrophages, smooth muscle cells (SMCs) and collagen. To evaluate macrophage and SMC content, we performed immunohistochemical analysis using antibodies against MAC-2 and α-actin, respectively. We found increased macrophage numbers in plaques (Figures 4 Ab and 4 Bb). There was no obvious difference in SMC number with PM2.5 administration (Figures 4 Ac and 4 Bc), compared with apoE-/- mice in the control group. Accordingly, the collagen content was also reduced in plaques, as shown by Sirius red staining (Figures 4 Ad and 4 Bd). These results suggest that PM2.5 administration was unfavorable for controlling atherosclerotic stability.
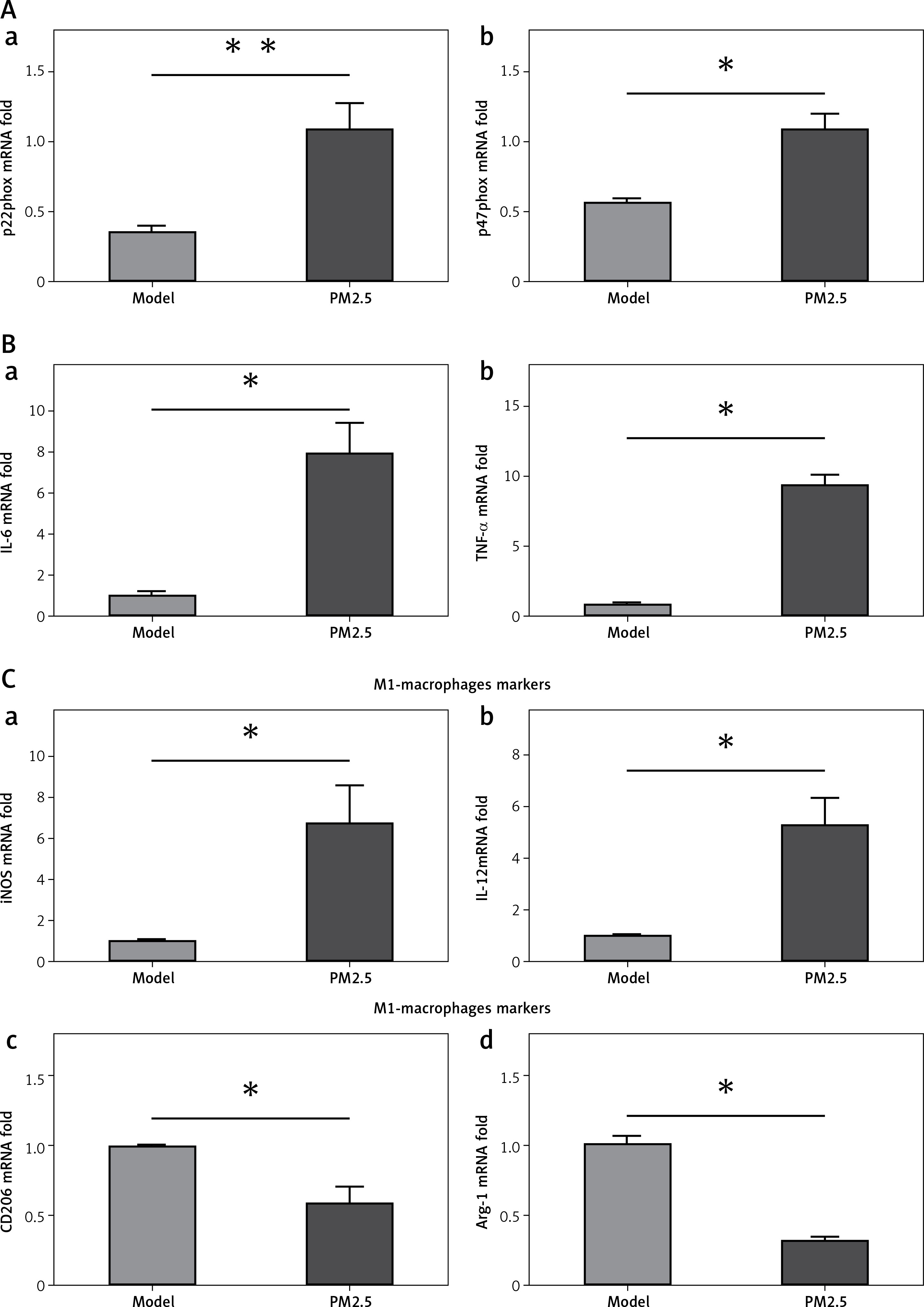
PM2.5 promoted M1 skewing and release of inflammatory factors by activated macrophages and platelets
Atherosclerosis is a chronic inflammatory disease in which pro-inflammatory factors amplify leukocyte recruitment and inflammatory responses via atherosclerosis-promoting cytokines. Circulating monocytes differentiate into macrophages in atherosclerotic plaques. Macrophages adapt their activation status or phenotype to mount appropriate responses, classified generally as M1 inflammatory and M2 immune regulating subtypes. M1 macrophages synthesize pro-inflammatory factors promoting atherosclerosis progression. M2 macrophages mediate anti-inflammatory and repair functions to inhibit atherosclerosis propagation. We found that classical pro-inflammatory cytokines, IL-6 and TNF-α, were significantly increased, along with increased M1-associated factors, iNOS and IL-12. Simultaneously, we observed decreased M2-specific factors – CD206 and arginase-1 – in plaques of descending arteries. These data suggested that PM2.5 administration increased inflammatory cytokine release and skewed macrophages toward the M1 phenotype, thus facilitating atherosclerosis progression (Figure 5 Ba, b and Ca, b).
Figure 5
PM2.5 treatment increased the expression of pro-inflammatory factors and M1-associated genes in apoE–/– mice. NAD(P)H oxidase subunits p22phox and p47phox expression (Aa, b, p = 0.0079 and 0.0294) and inflammatory cytokines IL-6 and TNF-α (Ba, b, p = 0.0291 and 0.0286) and M1-associated factors iNOS and IL-12 (Ca, b, p = 0.0122 and 0.0280) as well as M2-specific factors CD206 and arginase-1 (Cc, d, p = 0.0317 and 0.0216) from lesions of descending arteries were tested by the Q-PCR method
Data represent the mean ± SE.*P < 0.05 versus control.
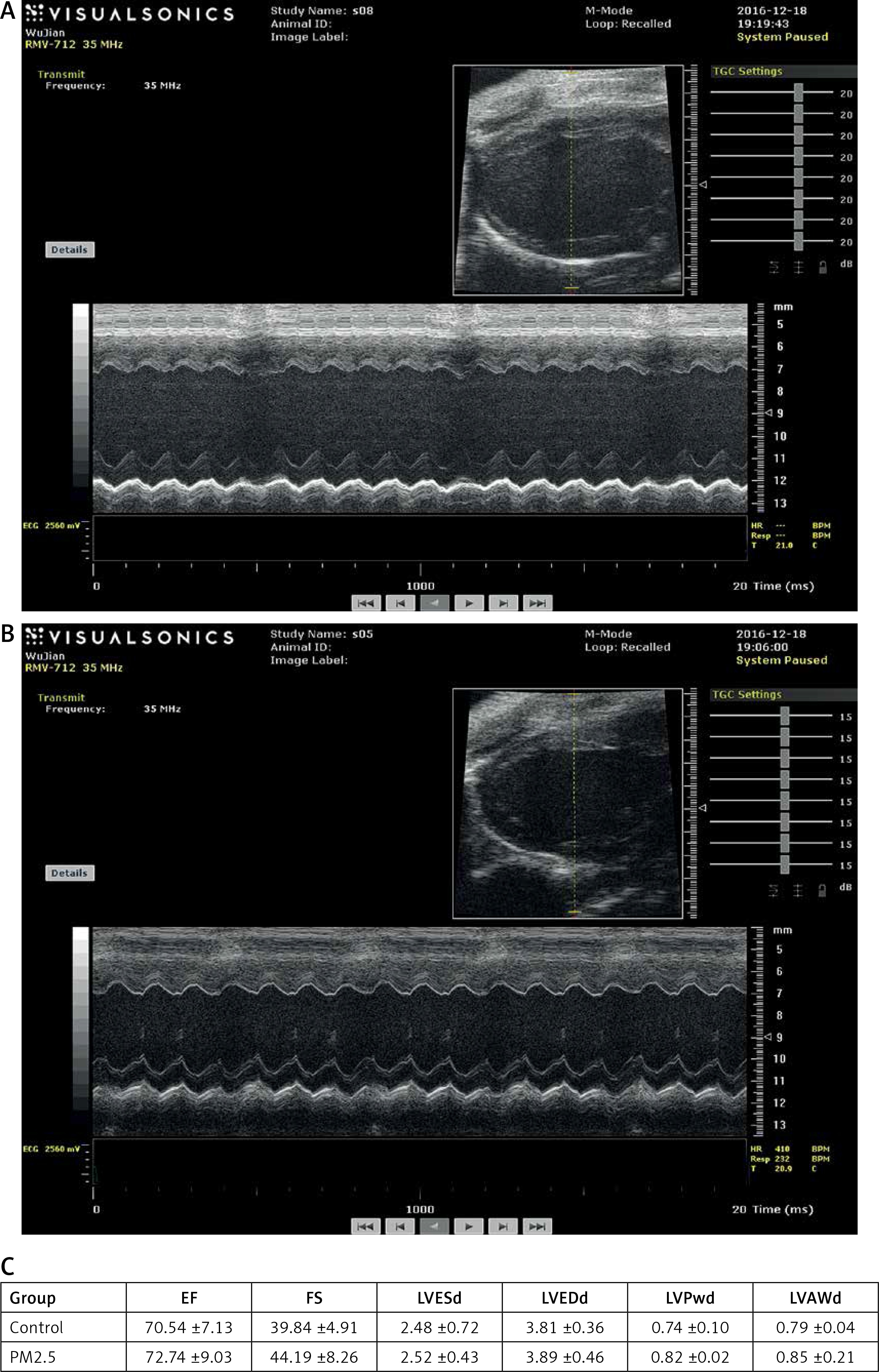
PM2.5 failed to affect cardiac function as measured by echocardiography
Quantitative analysis indicated that there were no differences in LVEDd and LVESd and no differences in LVAWd and LVESd between the groups, suggesting that PM2.5 did not affect the degree of myocardial hypertrophy, and the remaining indices were similar to those of control mice. All data are shown in Figure 6 (Tables III, IV).
Table III
Gene primer pairs
Table IV
Reagent information
Figure 6
PM2.5 failed to affect cardiac function tested by echocardiographic images in apoE–/– mice. A – Representative control group apoE–/– mice, heart structure; B – representative PM2.5 group, mice exposed to 30 mg/kg/day, LVAW and LVPW showed no significant difference compared to control mice. Data are shown in Figure 6 C
Discussion
We found that PM2.5 exposure accelerated atherosclerotic progression in apoE–/– mice. We found that apoE–/– mice instilled with pure PM2.5 showed: (1) significantly elevated serum pro-inflammatory factors levels, but caused no differences in serum glucose or lipids and failed to affect cardiac function tested by echocardiography; (2) more leukocyte and platelet accumulation on the surface of atheroma observed by SEM; (3) substantial endothelial injury, with greater numbers of platelets and leukocytes adhered; (4) greater lesion areas accompanied by greater numbers of accumulated macrophages, consistent with fewer SMCs and collagen content; and (5) strengthened pro-inflammatory cytokine secretion and lesion-associated macrophages, skewing to the M1 phenotype, with suppression of M2-associated cytokines as measured by Q-PCR. PM2.5 appeared to accelerate atherosclerotic progression primarily by activating leukocytes and platelets, as well as by activating inflammation and oxidative stresses in atherosclerotic plaques by switching macrophages to the M1 phenotype in apoE–/– mice.
Much epidemiological and experimental study of the fine particulate PM2.5 suggests that it is a risk factor for atherosclerotic progression, and therefore has acquired considerable attention worldwide. PM2.5 exerts atherogenic effects by inducing endothelial damage, mitochondrial injury, genotoxicity, inflammatory responses and oxidative stress [14]. Furthermore, many toxic compounds are adsorbed to fine particles in air pollutants, especially PM2.5. This agent penetrates the air-blood barrier, thereby damaging the respiratory, cardiovascular, and immune systems. Most developing countries are plagued by both serious atmospheric pollution and atherosclerosis-related diseases; therefore, a new mechanism of this association should be explored.
Atherosclerosis is a chronic inflammatory disease. Atherogenic factors are stimuli that trigger innate immunity and downstream inflammatory reactions. Circulating leukocytes, mainly monocytes and macrophages, are major components of innate immunity that act as the first defense line against foreign bodies [6, 7, 15–17]. Hyperlipidemia and hyperglycemia are classic triggers of atherosclerosis. Nevertheless, we found no significant difference between experimental and control groups with respect to glucose or lipids (Figure 1 A). However, we found elevated TNF-α, and IL-6 levels, both of which are associated with chronic inflammation [18–20]. These data suggest that PM2.5 promoted inflammatory responses in apoE–/– mice under atherosclerotic conditions, independent of an effect on lipid levels. In atherogenic initiation and progression, endothelial barriers break down [8] and platelets are almost immediately recruited. They adhere to endothelia and further express adhesion factors and receptors, including P-selectin, glycoprotein Ib, VI, GPIIb/IIIa and CD40L. The cascade leads to further accumulation and activation of leukocytes, especially monocytes [6, 7, 9]. We found that brachiocephalic arteries (BCA) suffered endothelial damage following PM2.5 treatment. The endothelial surface became rough (Figure 2 D, red arrows), with abundant platelets (Figures 2 C and F yellow arrows), and greater numbers of leukocytes (Figures 2 B, E, blue arrows) adhered on injured surfaces. These data suggested that PM2.5 augmented systemic inflammation and innate immune responses, at least partly by increasing the number of leukocytes and platelets as well as by promoting pro-inflammatory factor secretion.
In the pathophysiology of atherosclerosis, circulating modified lipoproteins are deposited on atheroma-prone arteries. Immune responses involving leukocytes and platelets have evolved to eliminate these foreign materials by initiating acute inflammatory reactions. However, such inflammation may become persistent and chronic under sustained atherogenic stimulus [10, 21]. We evaluated atherosclerotic areas of whole arteries by staining with Oil Red O. PM2.5 treatment increased red-positive areas (Figures 3 A, B).
Under pro-inflammatory conditions, circulating leukocytes transmigrate to atherosclerotic lesions and irreversibly differentiate into macrophages [11, 22]. However, macrophages have a high degree of plasticity, generally divided into two phenotypes. The first, classically activated macrophages (M1), synthesize abundant reactive oxygen species as well as pro-inflammatory cytokines. NADPH oxidases, especially nox2, generate superoxide in monocytes, producing ROS, thereby acting as a primary cause of atheroma. These stimulate leukocyte recruitment, activation, differentiation and survival. Among these, p22phox, p47phox, TNF-α, IL-6, IL-1β and IFN-α are associated with atherosclerosis [8, 10, 11, 23]. Macrophages of the second phenotype, alternatively activated macrophages (M2), secrete anti-inflammatory factors, including IL-10, TGF-β, arginase-1 and CD206, all of which exert anti-atherogenic effects [10, 11, 22, 24]. Therefore, the ratio of M1 to M2 macrophages in plaques is critical for development of atherosclerosis. Fibrous caps, mainly composed of collagen, maintain plaque integrity and prevent exposure of thrombotic necrotic cores to the circulation. SMCs are the major cellular component responsible for the synthesis of extracellular matrix [25], mainly collagen, and for maintaining rigid intra-plaque structures resistant to blood pressure [26–28]. M2 macrophages contribute to the proliferation and differentiation of medial SMCs and bone marrow fibrocytes. We found that the nox2-derived ROS sources p22phox and p47phox (Figures 5 Aa and b) and pro-inflammatory factors IL-6 and TNF-α, as well as M1-associated cytokines iNOS and IL-12 (Figures 5 Ba and b), were substantially elevated in the arteries of apoE–/– mice treated with PM2.5. There was a simultaneous decrease in the M2-specific markers arginase-1 and CD206 (Figures 5 B), indicating amplified pro-inflammatory effects of PM2.5 in atherosclerosis. The suppression of M2-switching could be at least partially explained by reduced SMCs and collagen content in atherosclerotic plaques of aortic roots (Figures 4 Bc and d).
In conclusion, we demonstrated the impact of PM2.5 exposure on the progression of atherosclerosis in a classical mouse model of cardiovascular disease. Our evidence suggested elevated amounts of platelets, increased inflammation and skewing toward a M1 subtype, being possible mechanisms of destabilization of atherosclerotic plaques. However, one should consider that the concentration of PM2.5 and the route of PM2.5 ingestion were different from real-life situations. This study was an animal study attempting to maximally imitate the harmful effects of PM2.5 on organisms.