Current issue
Archive
Manuscripts accepted
About the Journal
Editorial office
Editorial board
Abstracting and indexing
Subscription
Contact
Ethical standards and procedures
Most read articles
Instructions for authors
Article Processing Charge (APC)
Regulations of paying article processing charge (APC)
ONCOLOGY / BASIC RESEARCH
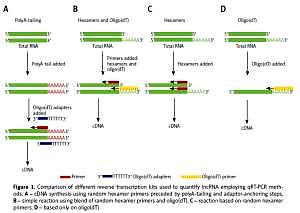
Quantification of long non-coding RNAs using qRT-PCR: comparison of different cDNA synthesis methods and RNA stability
1
Chair of Medical Biotechnology, Poznan University of Medical Sciences, Poznan,
Poland
2
Laboratory of Cancer Genetics, Greater Poland Cancer Centre, Poznan, Poland
3
Postgraduate School of Molecular Medicine, Medical University of Warsaw, Warsaw,
Poland
4
Department of Medical and Experimental Oncology, Heliodor Swiecicki Clinical
Hospital, Poznan University of Medical Sciences, Poznan, Poland
5
Department of Biology and Environmental Sciences, Poznan University of Medical
Sciences, Poznan, Poland
6
Department of Diagnostics and Cancer Immunology, Greater Poland Cancer Centre,
Poznan, Poland
Submission date: 2018-03-07
Final revision date: 2018-05-13
Acceptance date: 2018-05-29
Online publication date: 2019-01-30
Publication date: 2021-07-16
Arch Med Sci 2021;17(4):1006-1015
KEYWORDS
TOPICS
ABSTRACT
Introduction:
Long non-coding RNAs (lncRNAs), a class of regulatory RNA molecules, are over 200 nucleotides long and could be used as a new potential biomarker, but their detection methods such as qRT-PCR are still not validated, and the influence of RNA degradation on lncRNA quantification is not clear. In this study, commercially available cDNA synthesis kits were tested and the influence of RNA degradation was compared.
Material and methods:
Total RNA from FaDu cells was isolated and high quality RNA and highly degraded RNA samples were used. Reverse transcription was performed using three different commercially available kits and quantifications were performed using lncRNA Primer Plate and SYBR Green I Master by LightCycler 96. qRT-PCR was performed using three different cDNA samples and results are presented as the mean Ct values. A p-value < 0.05 was considered to be significant.
Results:
Lower lncRNA Ct values (61/90; 67.78%) after qRT-PCR quantification were observed for cDNA synthesized using random hexamer primers preceded by polyA-tailing and adaptor-anchoring steps. It was observed that 9/90 (10.00%) lncRNAs were not detectable using different cDNA synthesis methods. For 75/90 (83%) lncRNAs, RNA degradation weakly influenced lncRNA Ct values and no differences were observed between high quality RNA and degraded samples. Seventy percent of examined lncRNAs showed significantly different Ct values depending on RNA degradation.
Conclusions:
cDNA synthesis kits with random hexamer primers preceded by polyA-tailing and adaptor-anchoring steps allows enhancement of lncRNA quantification specificity and sensitivity. In most cases degradation of RNA samples does not affect lncRNA quantification because these molecules have good stability.
Long non-coding RNAs (lncRNAs), a class of regulatory RNA molecules, are over 200 nucleotides long and could be used as a new potential biomarker, but their detection methods such as qRT-PCR are still not validated, and the influence of RNA degradation on lncRNA quantification is not clear. In this study, commercially available cDNA synthesis kits were tested and the influence of RNA degradation was compared.
Material and methods:
Total RNA from FaDu cells was isolated and high quality RNA and highly degraded RNA samples were used. Reverse transcription was performed using three different commercially available kits and quantifications were performed using lncRNA Primer Plate and SYBR Green I Master by LightCycler 96. qRT-PCR was performed using three different cDNA samples and results are presented as the mean Ct values. A p-value < 0.05 was considered to be significant.
Results:
Lower lncRNA Ct values (61/90; 67.78%) after qRT-PCR quantification were observed for cDNA synthesized using random hexamer primers preceded by polyA-tailing and adaptor-anchoring steps. It was observed that 9/90 (10.00%) lncRNAs were not detectable using different cDNA synthesis methods. For 75/90 (83%) lncRNAs, RNA degradation weakly influenced lncRNA Ct values and no differences were observed between high quality RNA and degraded samples. Seventy percent of examined lncRNAs showed significantly different Ct values depending on RNA degradation.
Conclusions:
cDNA synthesis kits with random hexamer primers preceded by polyA-tailing and adaptor-anchoring steps allows enhancement of lncRNA quantification specificity and sensitivity. In most cases degradation of RNA samples does not affect lncRNA quantification because these molecules have good stability.
| eISSN: | 1896-9151 |
| ISSN: | 1734-1922 |
We process personal data collected when visiting the website. The function of obtaining information about users and their behavior is carried out by voluntarily entered information in forms and saving cookies in end devices. Data, including cookies, are used to provide services, improve the user experience and to analyze the traffic in accordance with the Privacy policy. Data are also collected and processed by Google Analytics tool (more).
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.