
Introduction
Vascular endothelial cells (VECs), as critical components of the inner lining of blood vessels, play a central role in maintaining systemic homeostasis [1]. During inflammatory responses, VECs are not merely passive responders but become activated and actively participate in the process [2]. By expressing adhesion molecules, selectins, chemokines, and other mediators, they effectively guide immune cells to target sites [3]. Some VECs further exhibit antigen-presenting cell-like functions, assuming a distinct role in humoral immune responses [4]. Meanwhile, persistent inflammatory stimuli drive the survival, proliferation, and migration of VECs through proangiogenic mechanisms, thereby promoting vascular remodelling and related pathological progression [5]. Together, these multifaceted regulatory mechanisms highlight the pivotal role of vascular endothelial cells in the initiation and progression of chronic inflammatory diseases.
Psoriasis is a chronic inflammatory skin disease mediated by abnormalities in the immune system, with a global prevalence of approximately 2–3% [6]. Its characteristic pathological features include dermal vascular abnormalities. Our previous studies have confirmed that psoriatic inflammatory stimuli can lead to vascular barrier disruption and increased permeability [7]. In this process, dysfunctional dermal VECs are activated and actively participate in disease progression by recruiting neutrophils and T cells into skin tissues, thereby modulating local immune responses [8, 9]. Diabetes, a common comorbidity of psoriasis, is a chronic metabolic disorder characterized primarily by persistent hyperglycaemia resulting from defects in insulin secretion or action [10]. Microvascular endothelial dysfunction constitutes a critical pathological aspect in its progression [11]. Recent epidemiological studies have shown that psoriasis patients have a significantly elevated risk of developing diabetes, while diabetes is also considered a potential risk factor for exacerbating psoriasis [12]. Moreover, patients with both psoriasis and diabetes are more prone to various vascular complications [13].
Although both diseases exhibit significant vascular endothelial dysfunction, the specific mechanisms linking psoriasis and diabetes remain unclear. Therefore, this study employs single-cell transcriptome analysis and weighted gene co-expression network analysis (WGCNA) to systematically investigate the potential shared pathogenesis between psoriasis and diabetes from the perspective of VEC dysfunction, identify common key genes, and further explore the shared pathways and genetic basis between psoriasis and diabetic foot ulcer (DFU) – a microvascular complication of diabetes – to uncover the potential molecular mechanisms underlying the increased risk.
Aim
This study aims to elucidate the shared pathogenesis between psoriasis and diabetes from the perspective of VEC dysfunction, identify common key genes, and explore their association with DFU through single-cell transcriptome analyses.
Material and methods
Data preprocessing and cluster annotation
This study acquired 10 single-cell RNA sequencing datasets from public database (GSE158924, GSE162183, GSE173706, GSE175990, GSE221648, PRJCA006797, PRJCA002692, E-MTAB-8142, PRJCA007038, GSE165816), comprising skin samples from four categories: healthy controls, psoriasis, diabetes, and DFU patients. Data preprocessing was performed using the Seurat package (v4.3.0) in R (v4.1.3) to complete the standard analytical pipeline [14]. During the quality control phase, we first filtered out cells with fewer than 3 expressed genes and those with a total gene count below 200. Subsequently, the Scater (v1.22.0) was employed to comprehensively evaluate cell counts and the proportion of mitochondrial/ribosomal genes, ensuring that subsequent analyses were based on high-quality cellular data. By identifying cells with a high expression of endothelial markers such as PECAM and CDH5, while excluding those expressing lymphatic endothelial signature genes like PROX1 and LYVE1, we successfully isolated the target VEC population.
To address batch effects during multi-dataset integration, we utilized the Harmony (v0.1.1) to integrate and correct VECs from different groups [15]. Based on the established VEC subtype classification system (cluster A, cluster C1, cluster C2, cluster P and cluster V) from previous research [16], we employed the FindTransferAnchors, TransferData, and MapQuery modules within the Seurat package to achieve systematic annotation of cell types.
Analysis of differentially expressed genes
To identify disease-specific expression profiles, we first performed differential gene analysis across the four clinical groups (healthy controls, psoriasis, diabetes, and DFU) using the FindMarkers function in Seurat (logfc.threshold = 0.25). Subsequently, Gene Ontology (GO) and KEGG pathway enrichment analyses were conducted on the identified differentially expressed genes using the ClusterProfiler package (v4.8.2) to elucidate their potential biological functions [17].
High-dimensional Weighted Gene Co-expression Network Analysis (hdWGCNA)
The hdWGCNA approach (v0.1.1.9010) [18] was employed to systematically identify key regulatory modules in psoriasis and diabetes progression. Specifically, gene expression matrices were first extracted from Seurat objects using the SetupForWGCNA function, followed by construction of representative metacells through the MetacellsByGroups function (Set/by setting the number of neighbouring cells to 50, and the maximum number of shared cells between individual cells to 10). After normalization and standardization pre-processing of the expression matrices, the SetDatExpr function was applied to define the analytical dataset. The TestSoftPowers function was then utilized to evaluate network topology and determine the optimal soft-thresholding power. Based on this parameter, a gene co-expression network was constructed using the ConstructNetwork function, and module eigengenes representing core features of each module were extracted via the ModuleEigengenes method. Ultimately, we identified 9 and 10 significant co-expression modules in the psoriasis and diabetes datasets, respectively, and then assessed their correlations with clinical traits using the ModuleTraitCorrelation function.
Construction of the protein-protein interaction (PPI) network
To further investigate functional regulatory relationships among proteins, we systematically constructed a PPI network based on previously co-expressed gene lists. The target gene set was first imported into the STRING database (https://string-db.org/), which leverages integrated multi-species protein interaction data to predict potential functional associations [19]. An interaction confidence threshold of > 0.4 was applied for preliminary filtering to enhance network reliability. The resulting interaction data were then imported into the Cytoscape platform (v3.9.1) for network visualization [20]. Finally, the CytoHubba was used to identify the top 10 hub nodes with the highest connectivity in the network, which may represent key proteins in disease-related pathways.
Cell culture
The human microvascular endothelial cell line (HMEC-1, CRL-3243) was procured from the American Type Culture Collection (ATCC, USA) and cultured following standard protocols. To simulate disease conditions, cells were seeded in 6-well plates and allowed to reach 70% confluence before being subjected to the following treatments: one group was exposed to psoriasis-like stimuli (IL-17A, TNF-α, and IFN-γ at concentrations of 50 ng/ml, 50 ng/ml, and 20 ng/ml, respectively) for 12 h, while another group was cultured under high-glucose conditions (30 mmol/l glucose) for 48 h. Following treatment, the cells were washed with PBS and collected for subsequent analysis.
qRT-PCR analysis
Total RNA was extracted with TRIzol Reagent (Invitrogen, Carlsbad, USA) and subsequently reverse transcribed into cDNA using the cDNA Synthesis kit (Bio-Rad Laboratories, USA). Quantitative real-time PCR (qRT-PCR) was carried out on the CFX384 system (Bio-Rad Laboratories, USA) using the synthesized cDNA as template, along with gene-specific primers (Supplementary Table S1) and SYBR Green Supermix (Bio-Rad Laboratories, USA). The thermal cycling protocol consisted of an initial denaturation at 95°C for 10 min, followed by 40 cycles of amplification, each comprising denaturation at 95°C for 10 s and combined annealing/extension at 60°C for 30 s. Fluorescence data were collected and analysed with CFX Manager software (Bio-Rad Laboratories, USA) based on the comparative threshold cycle method. The relative expression levels of target mRNAs were determined after normalization to the internal reference gene β-actin.
Data visualization
Visualization of the single-cell dataset was performed in the R environment using multiple specialized packages, with uniform parameter settings applied across comparative datasets. Specifically, ggplot2 (v3.5.1) was employed for generating fundamental statistical graphics, ggvenn (v0.1.9) for constructing Venn diagrams, and ggalluvial (v0.12.5) for creating alluvial diagrams to visualize categorical data flow.
Statistical analysis
All experiments were independently replicated more than three times. Data are expressed as mean ± standard deviation. Statistical evaluations were conducted with GraphPad Prism software (v9.0.0), using one-way ANOVA as appropriate. Differences were considered statistically significant at p < 0.05.
Results
VECs can be classified into five subclusters
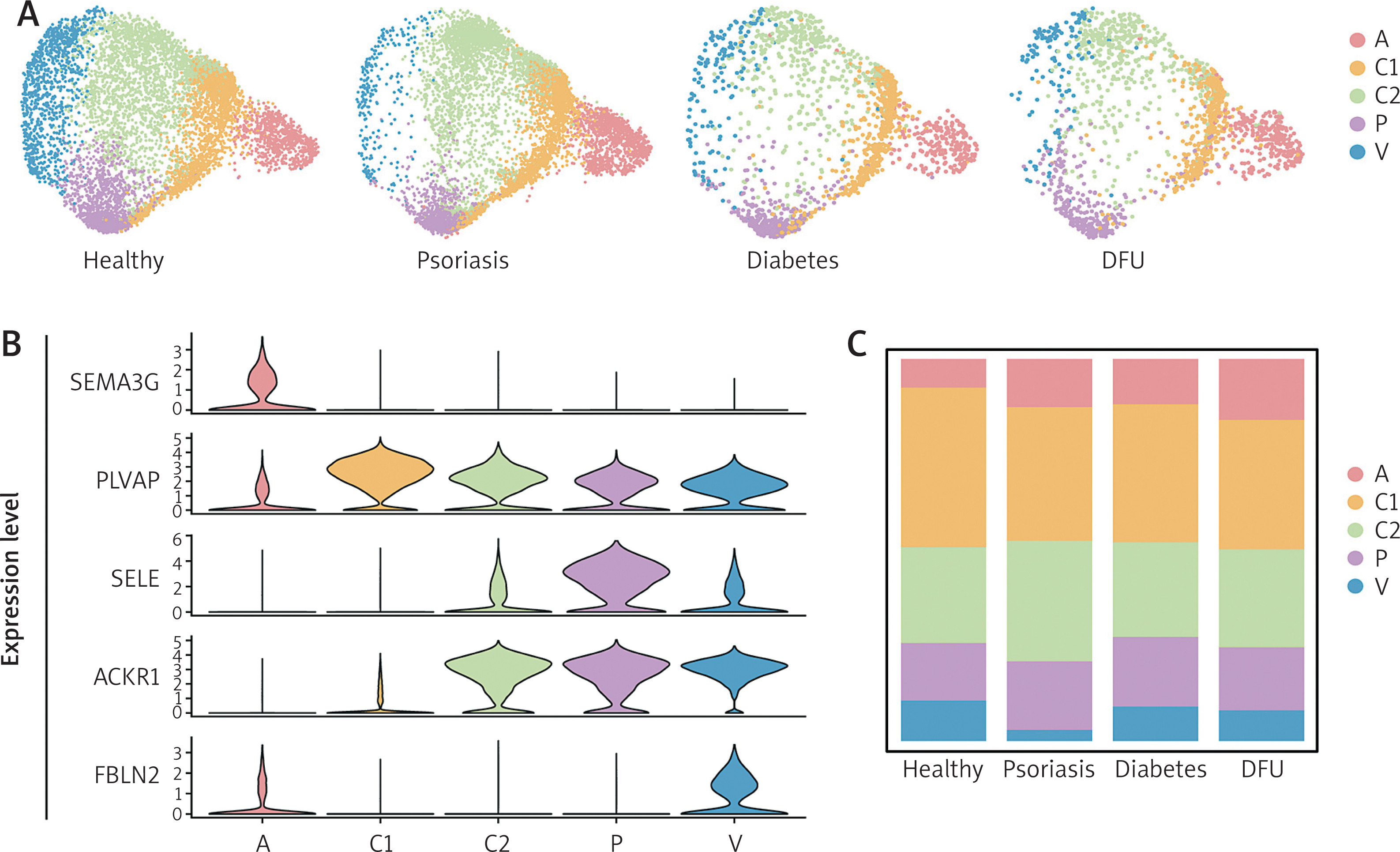
We conducted systematic identification of VECs based on single-cell data of skin tissues from public databases, including skin samples from 20 healthy individuals, 26 psoriasis patients, 7 diabetes patients and 5 DFU patients. VECs were extracted from these samples, and a total of 9890 cells from the healthy group, 9108 cells from the psoriasis group, 1840 cells from the diabetes group, and 1765 cells from DFU group were obtained for subsequent analysis. Based on an established reference atlas, we categorized VECs into five distinct clusters via cell identity transfer, including arteriole VECs (cluster A), capillary VECs (cluster C1; cluster C2); postcapillary venule VECs (cluster P), and venule VECs (cluster V) (Figure 1 A). Each cluster was distinguished by specific marker genes: SEMA3G, PLVAP, SELE, ACKR1, and FBLN2 (Figure 1 B). Cluster distribution analysis revealed that compared with the healthy control group, the proportions of cluster A and P were significantly elevated in psoriasis, diabetes, and DFU groups (Figure 1 C), suggesting that these two clusters may hold important pathological significance under disease conditions.
Figure 1
IscRNA-seq analysis of VECs in human skin from healthy, psoriasis, diabetes, and DFU groups. A – UMAP plot of human skin VECs clustered in five groups from four skin states. B – Violin Plots showing expressions of SEMA3G, PLVAP, SELE, ACKR1, FBLN2 in each subcluster. C – Barplot showing proportions of corresponding clusters across four skin conditions
Shared molecular networks in VECs of psoriasis and diabetes
Differential gene expression analysis revealed that VECs from psoriasis patients showed significant enrichment in pathways related to inflammatory immune responses, cell adhesion and migration, angiogenesis, and metabolic dysregulation (Figure 2 A). In contrast, VECs from diabetic patients primarily exhibited activation of pathways involved in metabolic disturbances, insulin resistance, vascular dysfunction, inflammatory immune responses, cell junction organisation, and cytoskeletal remodelling (Figure 2 B). Although both diseases involve inflammatory and metabolic abnormalities, their core pathological mechanisms exhibit significant differences: in psoriasis, VECs promote immune cell infiltration into skin sites and pathological angiogenesis, thereby supporting abnormal epidermal hyperplasia; whereas diabetic VECs are under sustained exposure to hyperglycaemia, AGE-RAGE signalling pathway activation, and insulin resistance, developing vasodilatory dysfunction and vascular structural remodelling, ultimately driving the occurrence of various vascular complications.
Figure 2
Common pathogenic pathways in VECs of psoriasis and diabetes. A – Dotplot showing enriched terms of VECs in psoriasis. The colour indicates the –log10 p-value, and the size indicates the count. B – Dotplot showing enriched terms of VECs in diabetes. The colour indicates the –log10 p-value, and the size indicates the count;. C – Venn diagram illustrating the number of upregulated common DEGs of VECs in psoriasis and diabetes. D – Venn diagram illustrating the number of downregulated common DEGs of VECs in psoriasis and diabetes. E – Dotplot showing enriched pathways of genes that are upregulated in both diabetic and psoriatic VECs. The colour indicates the –log10 p-value, and the size indicates the count
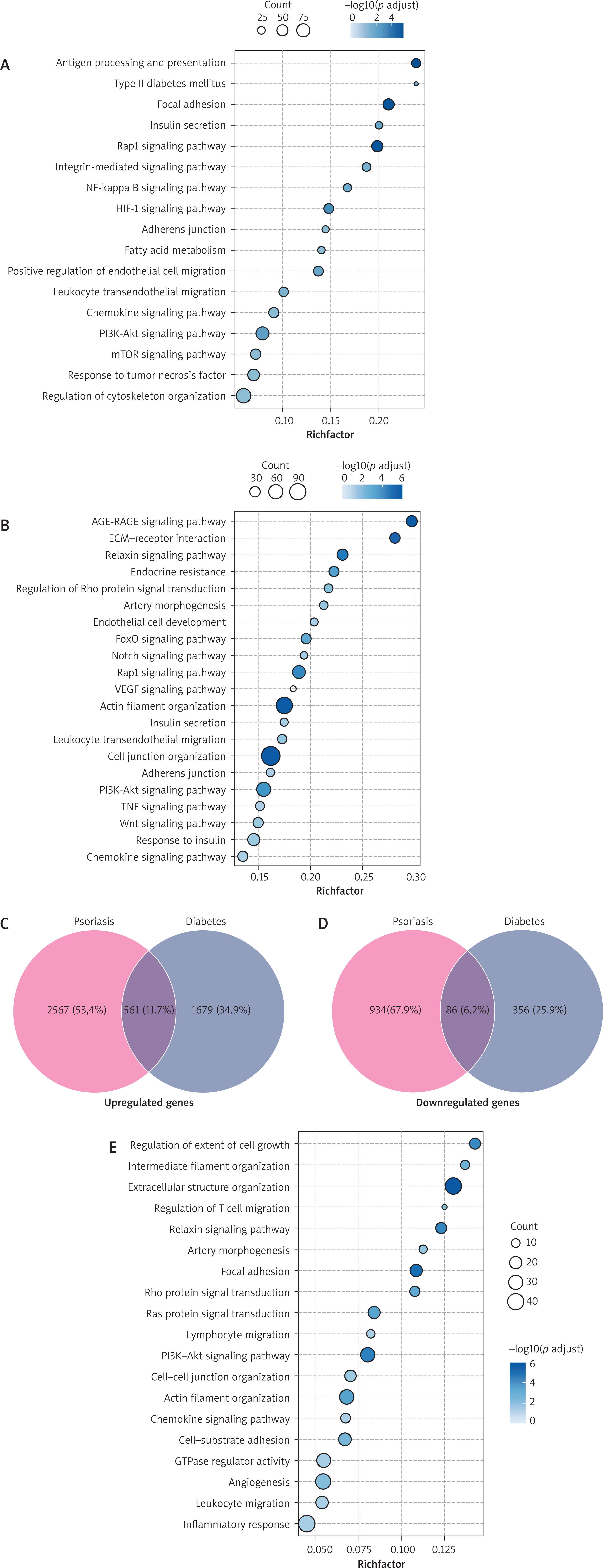
Further analysis revealed 561 commonly upregulated genes and 86 downregulated genes in both psoriasis and diabetes compared with healthy controls (Figures 2 C, D). These shared upregulated genes collectively establish a molecular framework common to both diseases, co-ordinately regulating several key biological processes: activating immune cell migration-related pathways (chemokine signalling, leukocyte transendothelial migration), modulating cell junction structure and cytoskeletal dynamics (intercellular junction assembly, Rho GTPase signalling), while simultaneously promoting pathological angiogenesis (PI3K-Akt signalling, angiogenesis regulation) (Figure 2 E). These processes together constitute the key shared mechanism of “inflammatory cell infiltration – vascular structural remodelling – vascular dysfunction” in psoriasis and diabetes.
Identification of core shared genes in VECs of psoriasis and diabetes
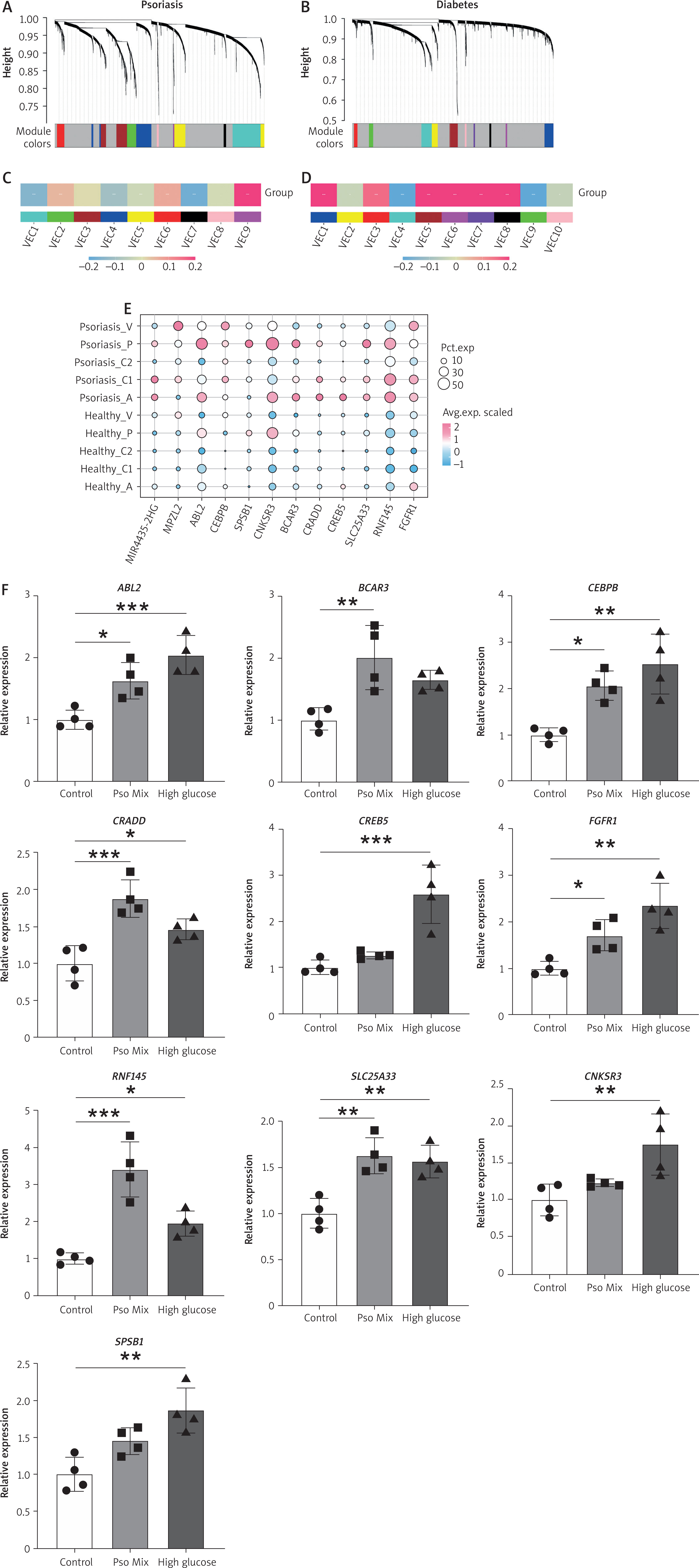
Gene co-expression networks were constructed from diabetic and psoriatic datasets using hdWGCNA (Figures 3 A, B). Specific module analysis revealed that VEC9 module showed a strong positive correlation with the disease in psoriasis, while VEC6 module exhibited the strongest positive correlation with the disease state in diabetes (Figures 3 C, D). Through an intersection analysis of high-correlation modules from psoriasis and diabetes, and differential expressed genes (DEGs) from both diseases compared to healthy controls, we ultimately identified 12 core genes (Figure 3 E). These genes form a molecular network that reveals the shared pathological mechanisms of both diseases: transcription factors CEBPB and CREB5 act as core regulatory elements, coordinating broad pro-inflammatory gene expression programs; FGFR1 and ABL2 significantly enhance inflammation and proliferation signalling through their kinase activities; SPSB1 and RNF145 precisely regulate the stability of key inflammatory and metabolic proteins via ubiquitination; BCAR3 and CNKSR3 integrate multiple pathway signals as signalling hubs, regulating cytoskeletal dynamics and adhesion; CRADD establishes the link between inflammation and cell death by mediating apoptotic signals; MPZL2 participates in cell junction and barrier function regulation; while SLC25A33 maintains mitochondrial metabolic substrate balance, providing energy and biosynthetic support for the inflammatory process. We examined the expression levels of the representative core genes under psoriatic stimulation and high-glucose conditions in HMEC cultures, and found that these genes showed varying degrees of increased expression in both conditions (Figure 3 F). Notably, these core genes are specifically expressed mainly in clusters A and P (Figure 3 E), suggesting their pivotal role in driving the comorbid progression of psoriasis and diabetes.
Figure 3
Core hub genes in VECs of psoriasis and diabetes. A – The cluster dendrogram of VEC genes in psoriasis. B – The cluster dendrogram of VEC genes in diabetes. C – Module-trait relationships in psoriasis. The colour indicates the correlation value. D – Module-trait relationships in diabetes. The colour indicates the correlation value. E – Dotplot showing overlapping genes between high-correlation modules from psoriasis and diabetes, and DEGs from both diseases compared to healthy controls. The colour indicates average expression of genes and the size indicates the percent expressed of genes. Significance was calculated using one-way ANOVA with Tukey’s post hoc test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 F – mRNA expression of representative core genes of HEMCs treated with psoriatic mix and high glucose. Significance was calculated using one-way ANOVA with Tukey’s post hoc test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001
Identification of DFU-associated genes in the context of psoriasis
Epidemiological evidence indicates that patients with coexisting psoriasis and diabetes have a significantly increased risk of developing DFU. Therefore, we sought to identify key genes associated with the development of DFU in psoriasis. We found that key genes in DFU were simultaneously enriched in immune activation and tissue remodelling compared to diabetes. On the one hand, humoral immune response and antibacterial defence pathways were significantly upregulated, suggesting the presence of a persistent and immuneinflammatory response at the wound site. Simultaneously, cytoskeleton-related genes and wound healing-related genes were highly active, reflecting active initiation of repair programs (Figure 4 A).
Figure 4
Shared genetic basis of psoriasis and DFU. A – Dotplot showing enriched terms of VECs in DFU compared to diabetes. The colour indicates the –log10 p-value, and the size indicates the count. B –Venn diagram illustrating the number of upregulated and downregulated common DEGs of VECs in psoriasis and DFU; C – Dotplot showing enriched pathways of genes that are upregulated and downregulated in both psoriatic and DFU VECs. The colour indicates the –log10 p-value, and the size indicates the count; D – The protein-protein interaction network constructed from upregulated genes in both psoriatic and DFU VECs. E – Dotplot showing expression of core genes selected by PPI network. The colour indicates average expression of genes and the size indicates the percent expressed of genes
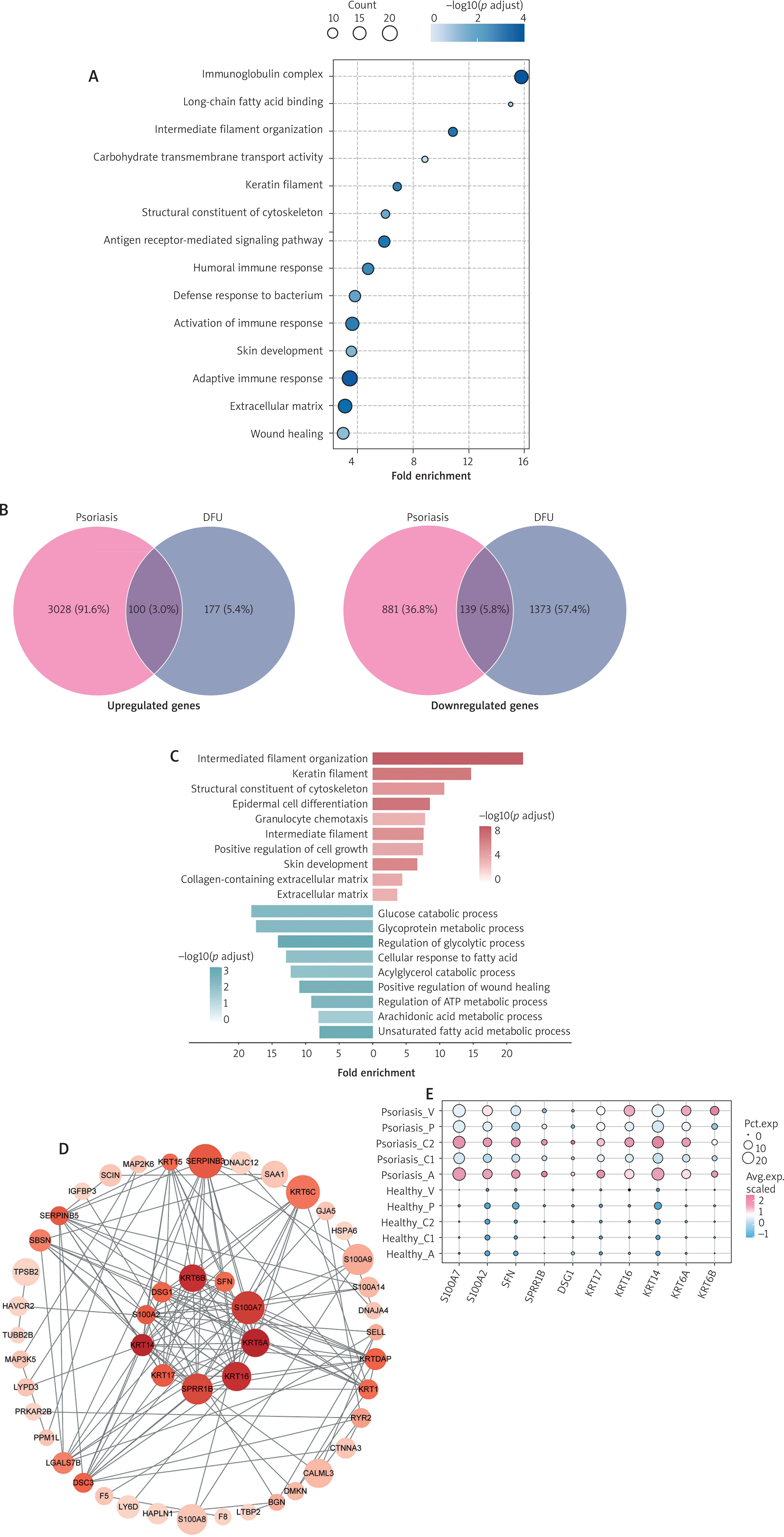
By conducting an intersection analysis of the differentially expressed genes between DFU and psoriasis, 100 upregulated genes and 139 downregulated genes were identified (Figure 4 B). Pathway analysis of the commonly upregulated genes revealed that VECs were in an abnormally activated state, with their characteristic gene expression profile driving cytoskeletal reorganisation and pathological angiogenesis. These alterations not only directly increased vascular permeability but also persistently amplified the inflammatory response through mechanisms such as promoting granulocyte chemotaxis. Among the downregulated genes, we observed disruptions in key metabolic processes, including glucose breakdown, ATP synthesis, and lipid metabolism, along with significant inactivation of wound healing-related mechanisms, leading to severely impaired abilities to maintain vascular homeostasis and initiate tissue repair (Figure 4 C).
Further PPI network analysis identified core genes co-regulated in both DFU and psoriasis (Figure 4 D). Among the upregulated genes, key molecules such as S100A7, S100A2, KRT16 and KRT17 collectively formed a pathological network centred on inflammatory response and barrier dysfunction. Mechanistically, the S100 protein family, as important inflammatory mediators, continuously promoted a chronic inflammatory cycle, while high expression of keratin family genes led to cytoskeletal reorganisation and compromised endothelial barrier integrity. Notably, these genes were specifically highly expressed in clusters A and C2, suggesting that these two clusters may play key roles in the pathological process of DFU (Figure 4 E).
Discussion
Diabetes is recognized as a common comorbidity of psoriasis, with evidence establishing psoriasis as an independent risk factor for insulin resistance [21]. VECs are not only involved in the pathogenesis of psoriasis but are also closely associated with characteristic vascular complications of diabetes, suggesting that these conditions may share an interconnected mechanism centred on endothelial dysfunction. Through transcriptomic analysis of VECs in psoriasis and diabetes, we identified 561 co-upregulated genes and further pinpointed 12 key genes.
CEBPB triggers inflammasome activation and exacerbates metabolic disturbances in diabetes, directly impairing the reparative capacity of endothelial progenitor cells and aggravating insulin resistance [22]. In psoriasis, CEBPB is also regarded as a critical biomarker, with its binding sites enriched in the promoter regions of target genes IL-17R [23]. ABL2 modulates Rho GTPase-mediated dynamics of cell adhesion, thereby compromising vascular barrier integrity and increasing permeability [24]. FGFR1 is activated by high glucose via the TLR4/c-Src pathway, leading to diabetic myocardial inflammatory responses [25]. This receptor is also involved in modulating the NF-κB signalling pathway, influencing the suppression of inflammatory responses. CRADD contributes to the maintenance of endothelial barrier integrity by inhibiting inflammatory factors such as IL-6 in VECs [26]. MPZL2 encodes an adhesion molecule that mediates inter-epithelial interactions [27] and has been identified as a potential molecular marker of early endothelial impairment in type 2 diabetes [28]. SLC25A33 drives mitochondrial DNA generation and reactive oxygen species accumulation, ultimately leading to aberrant vascular smooth muscle cell proliferation, migration, and pathological neointima formation [29].
DFU represents one of the most challenging complications of diabetes [30]. Patients with psoriasis exhibit an elevated risk of diabetes-related vascular complications, prompting us to analyse shared signalling pathways and genes in VECs between psoriasis and DFU. Among the upregulated key genes, the S100 family displayed prominent expression. Previous studies have demonstrated that in psoriasis, S100A7 binds to the RAGE receptor, inducing reactive oxygen species generation and upregulating vascular endothelial growth factor expression, thereby promoting endothelial cell proliferation and angiogenesis [31, 32]. S100A7 was also found to be significantly overexpressed in skin lesions of patients with DFU [33]. These findings suggest that S100A7 may play an important role in the sustained inflammatory responses common to both psoriasis and DFU. Besides, S100A2 was observed to colocalize with K14 in skin tissues, which is also a key gene commonly upregulated in both DFU and psoriasis. Studies indicate a positive correlation between S100A2 and K14 levels, and the expression level of S100A2 is closely related to the severity of the disease [34].
It is noteworthy that among the key genes commonly upregulated in both psoriasis and DFU, keratin molecules have significantly been identified. In psoriatic lesional skin, KRT6/16/17 overexpression is regarded as a characteristic biomarker of the disease [35–37]. Furthermore, they actively participate in regulating keratinocyte proliferation and inflammatory responses, thereby promoting the formation of psoriatic plaques. Their persistent overexpression is primarily regulated by T cell-derived cytokines, such as IL-17A, IFN-γ, and IL-22 [38–40]. Whereas during skin injury, the expression of KRT6/16/17 is also upregulated, which is mainly driven by damage-associated molecular patterns (DAMPs) released from necrotic cells via activation of the NF-κB/MAPK signalling pathway [41–43].
Conclusions
This study systematically elucidates the shared pathological mechanisms between psoriasis and diabetes at the VEC level. We identified functional commonalities in endothelial cells between psoriasis and diabetes and discovered 12 core hub genes, and clarified the critical roles of VEC clusters such as A and C2 in mediating vascular barrier disruption and pathological angiogenesis. Notably, the shared mechanisms between psoriasis and DFU may be collectively driven by sustained inflammatory responses mediated by the S100 protein family and tissue remodelling driven by the keratins, providing a molecular explanation for the increased susceptibility to DFU in patients with psoriasis and diabetes. These findings not only deepen the understanding of the comorbidity mechanisms between psoriasis and diabetes, but also provide a new theoretical basis and targets for developing common therapeutic strategies targeting VEC dysfunction.







