Current issue
Archive
Manuscripts accepted
About the Journal
Editorial office
Editorial board
Abstracting and indexing
Subscription
Contact
Ethical standards and procedures
Most read articles
Instructions for authors
Article Processing Charge (APC)
Regulations of paying article processing charge (APC)
PHARMACOLOGY AND PHARMACY / BASIC RESEARCH
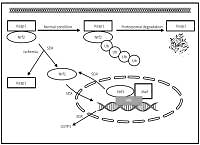
Sulodexide up-regulates glutathione S-transferase P1 by enhancing Nrf2 expression and translocation in human umbilical vein endothelial cells injured by oxygen glucose deprivation
1
Department of Pharmacology, School of Medicine, Medical University of Silesia, Katowice, Poland
2
Department of Internal Medicine and Clinical Pharmacology, School of Medicine, Medical University of Silesia, Katowice, Poland
3
Department of General Surgery, Vascular Surgery, Angiology and Phlebology, School of Medicine, Medical University of Silesia, Katowice, Poland
Submission date: 2018-12-01
Final revision date: 2018-12-26
Acceptance date: 2019-01-06
Online publication date: 2019-02-11
Publication date: 2020-05-26
Arch Med Sci 2020;16(4):957-963
KEYWORDS
TOPICS
ABSTRACT
Introduction:
Sulodexide (SDX) is used for the treatment of many vascular disorders due to its anticoagulant, anti-inflammatory and anti-atherosclerotic properties. However, the detailed molecular mechanism of its endothelioprotective action is still not completely understood. There is increasing evidence suggesting that antioxidant enzymes play an important role in anti-ischemic properties of SDX. We postulate that up-regulation of glutathione-S-transferase P1 (GSTP1) mediated by the transcription factor Nrf2 could be associated with the antioxidant effect of SDX on vascular endothelial cells.
Material and methods:
In the present study, we investigated whether SDX affects GSTP1 and Nrf2 in oxygen glucose deprivation (OGD) treated human umbilical vein endothelial cells (HUVECs). The cells treated with/without SDX (0.5 LRU/ml) were subjected to OGD for 1–6 h. To study the influence of SDX on the Nrf2 nucleus accumulation, the cells were incubated with 0.5 LRU/ml SDX in OGD for 1 h.
Results:
We found that after short-term OGD (1–3 h), the drug increased the expression of both GSTP1 and Nrf2 mRNA/protein in HUVECs (p < 0.05), as determined by real-time PCR and enzyme-linked immunosorbent assay (ELISA). SDX treatment also enhanced the nuclear accumulation of Nrf2 in HUVECs after 1 h of OGD (p < 0.05).
Conclusions:
SDX induces a rapid onset of the antioxidant response by up-regulating the expression of GSTP1 and Nrf2 in endothelial cells subjected to in vitro simulated ischemia.
Sulodexide (SDX) is used for the treatment of many vascular disorders due to its anticoagulant, anti-inflammatory and anti-atherosclerotic properties. However, the detailed molecular mechanism of its endothelioprotective action is still not completely understood. There is increasing evidence suggesting that antioxidant enzymes play an important role in anti-ischemic properties of SDX. We postulate that up-regulation of glutathione-S-transferase P1 (GSTP1) mediated by the transcription factor Nrf2 could be associated with the antioxidant effect of SDX on vascular endothelial cells.
Material and methods:
In the present study, we investigated whether SDX affects GSTP1 and Nrf2 in oxygen glucose deprivation (OGD) treated human umbilical vein endothelial cells (HUVECs). The cells treated with/without SDX (0.5 LRU/ml) were subjected to OGD for 1–6 h. To study the influence of SDX on the Nrf2 nucleus accumulation, the cells were incubated with 0.5 LRU/ml SDX in OGD for 1 h.
Results:
We found that after short-term OGD (1–3 h), the drug increased the expression of both GSTP1 and Nrf2 mRNA/protein in HUVECs (p < 0.05), as determined by real-time PCR and enzyme-linked immunosorbent assay (ELISA). SDX treatment also enhanced the nuclear accumulation of Nrf2 in HUVECs after 1 h of OGD (p < 0.05).
Conclusions:
SDX induces a rapid onset of the antioxidant response by up-regulating the expression of GSTP1 and Nrf2 in endothelial cells subjected to in vitro simulated ischemia.
Share
RELATED ARTICLE
| eISSN: | 1896-9151 |
| ISSN: | 1734-1922 |
We process personal data collected when visiting the website. The function of obtaining information about users and their behavior is carried out by voluntarily entered information in forms and saving cookies in end devices. Data, including cookies, are used to provide services, improve the user experience and to analyze the traffic in accordance with the Privacy policy. Data are also collected and processed by Google Analytics tool (more).
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.