Congenital adrenal hyperplasia (CAH) refers to a group of autosomal recessive disorders in steroid and glucocorticoid synthesis including cortisol and aldosterone, secondary to an enzymatic deficiency. In 21-hydroxylase deficiency (21-OHD), the most common form of CAH accounting for > 90% of cases, the impaired production of cortisol and aldosterone triggers an enzymatic cascade leading to overproduction of adrenocorticotropic hormone (ACTH), adrenal hyperplasia, and excess adrenal androgen production. In its most severe form, the classic salt wasting variety, patient presentation is usually in infancy with salt wasting and dehydration secondary to an Addisonian crisis. Female infants with the classic form present with ambiguous genitalia due to elevated adrenal androgens. A less severe form of classic CAH due to 21-OHD presents without salt wasting; male children with this present with signs of virilization in early childhood, if they had not been identified by neonatal screening. Milder cases of CAH due to 21-OHD, the late-onset (nonclassic) form, are much more common than the classic forms, and may present with a spectrum of clinical signs and symptoms including precocious puberty, hirsutism, and oligomenorrhea. Male patients with the classic forms frequently develop testicular adrenal rest tumors (TARTs), which are benign masses within the rete testes and frequently cause infertility. Conversely, those with the more common late-onset (non-classic) form rarely develop TARTs. TARTs may also develop in men with the much rarer CAH due to 11β-hydroxylase deficiency, which occurs in up to 5% of cases, and typically presents with hypertension, hypokalemia, and sexual precocity or early puberty. Other forms of CAH are exceedingly rare. Mainly, 17-OHD and combined 17-OHD/17-20 lyase deficiency can cause hypertension, hypokalemia, and abnormal sexual development.
A 33-year-old man presented to the endocrinology department with a chief complaint of leg cramps. The patient was diagnosed with congenital adrenal hyperplasia due to 21-OHD at age 2 weeks. He had been receiving steroid supplementation for the majority of his life, but admitted to poor compliance with medications and medical care for about 3 years mostly due to his tenuous health insurance situation. Four days before his appointment he developed leg cramps after vigorous exercise. He was married for 2 years and recently he and his wife had been trying to conceive, without success. He had no complaints of fatigue or sexual dysfunction. On physical examination he appeared healthy, well masculinized, but had slightly small, firm testes. Laboratory evaluation returned with very high levels of 17-hydroxyprogesterone and androstenedione, low morning cortisol, and low normal testosterone. He was started on dexamethasone 1 mg at bedtime and fludrocortisone daily and a scrotal ultrasound was ordered. He returned to the endocrinology clinic after 3 months with a 15-pound weight gain. He had not yet undergone the scrotal ultrasound. Because of the weight gain his glucocorticoid was changed to hydrocortisone three times daily with doses of 10 mg, 5 mg, and 5 mg. Six months later, on his next follow-up visit, a scrotal ultrasound was reordered.
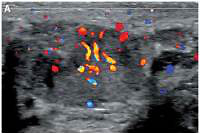
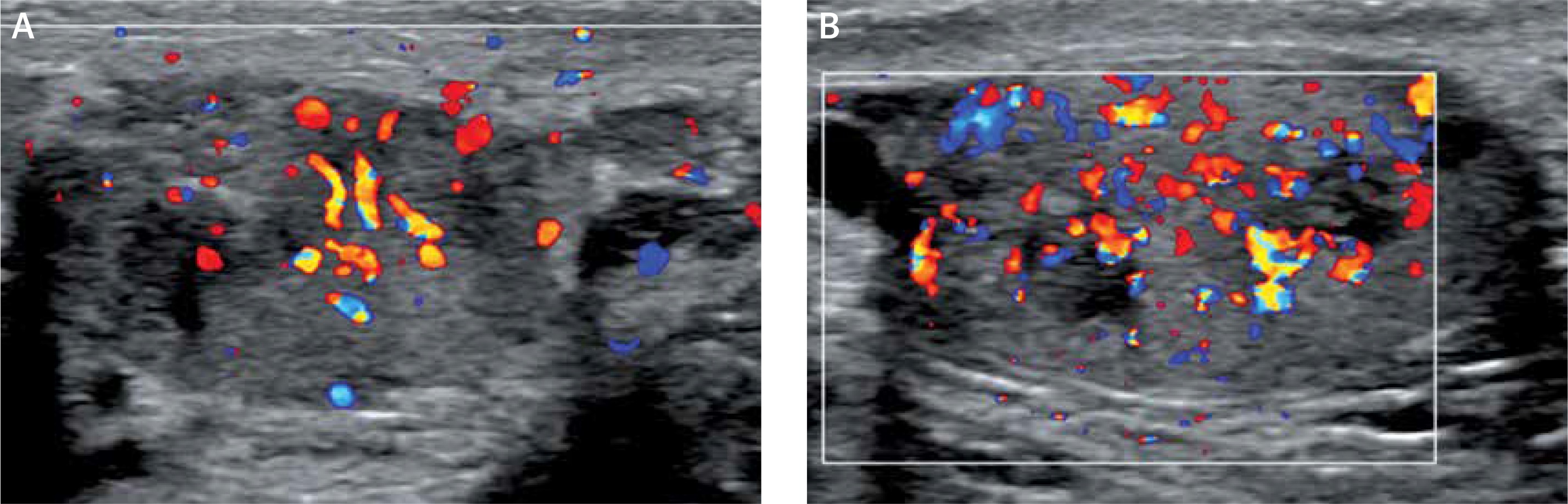
The initial scrotal ultrasound revealed single bilateral heterogeneously hypoechoic intratesticular masses with irregular borders. These masses occupied approximately 50% of the testicular parenchyma (Figure 1). Doppler interrogation showed extensive vascularity within both masses (Figure 2). Given the clinical history of congenital adrenal hyperplasia, the findings likely reflected bilateral adrenal rest tumors. The differential diagnostic considerations of lymphoma, metastatic disease, or sarcoidosis were considered less likely in this clinical setting. A semen analysis revealed azoospermia. A 6-month follow-up scrotal ultrasound after change of his glucocorticoid to dexamethasone 0.5 mg at bedtime daily demonstrated that the heterogeneous appearing bilateral testicular masses had minimally decreased in size (Figure 3). The vascularity within both masses was similar to slightly decreased in prominence (Figure 4).
Figure 1
Initial sonographic evaluation (November 2017) showed bilateral hypoechoic heterogeneous vascular intratesticular masses (A – right testicle, B – left testicle) with irregular borders extending to the epidydimal heads estimated to occupy approximately 50% of the testicular parenchyma
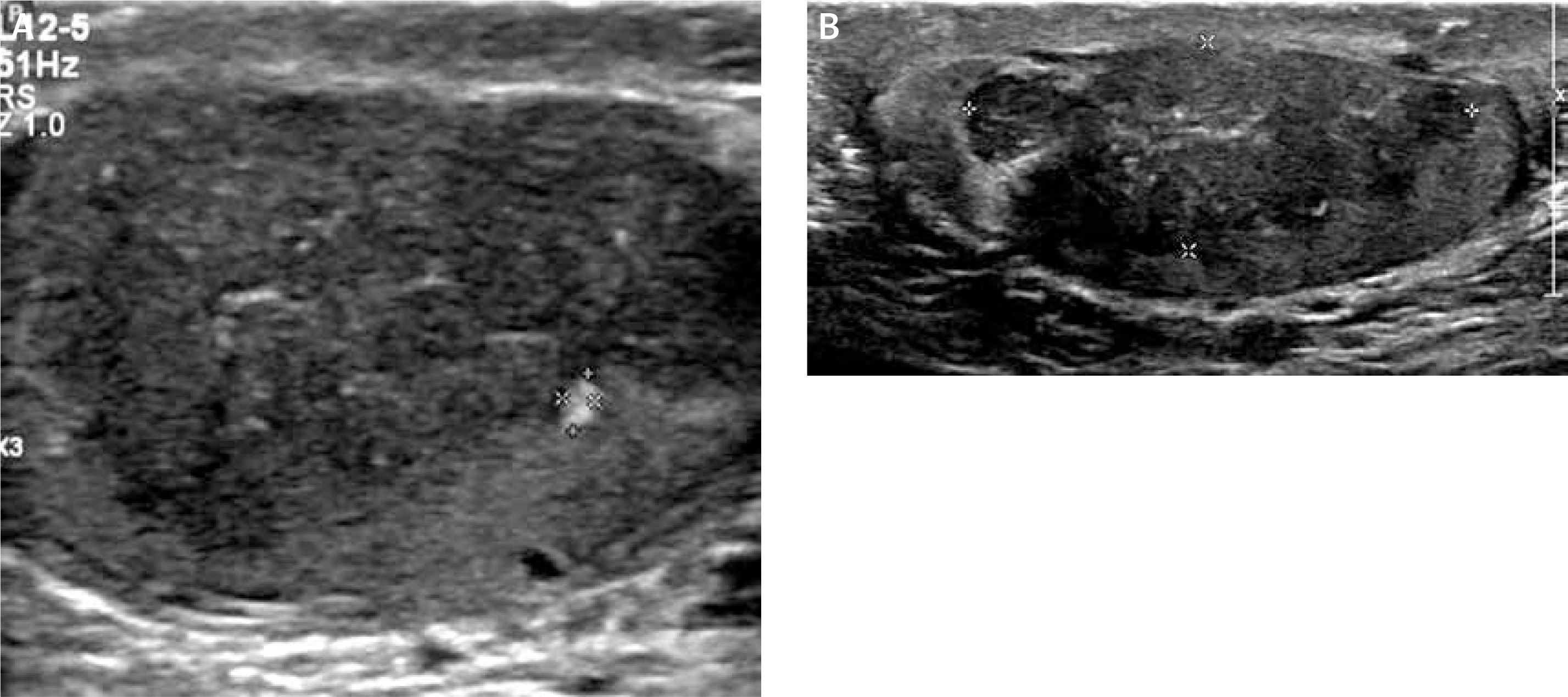
Figure 2
Doppler interrogation showed extensive vascularity within both masses (A – right testicle, B – left testicle)
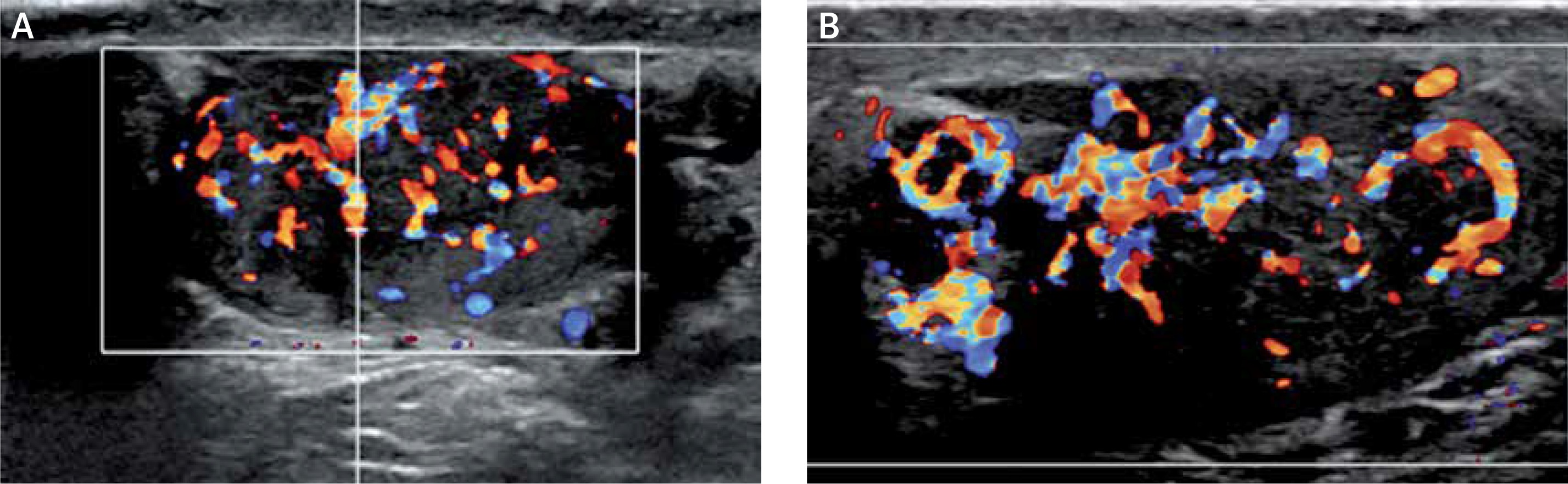
Figure 3
Six-month follow-up sonographic evaluation of both masses (April 2018) showed that glucocorticoid up-titration after initial ultrasound revealed that the testicular masses had minimally decreased in size (A – right testicle, B – left testicle)
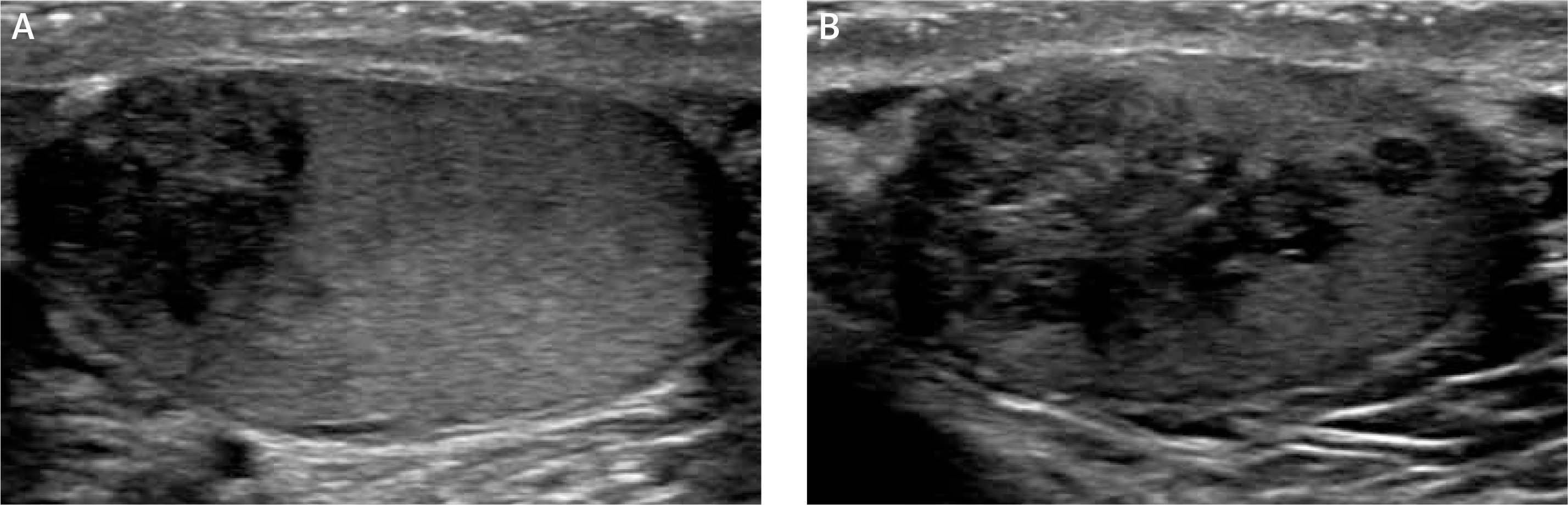
Figure 4
Internal vascularity of 6-month sonographic evaluation shows decreased vascularity (A – right testicle, B – left testicle)
TARTs were first described in the literature by Wilkins in 1940 [1]. These tumors are frequently found in young men with a history of CAH due to 21-OHD with a reported prevalence varying from 0 to 94% depending on patient selection, hormonal control, and imaging modality utilized for diagnosis [2, 3]. The etiology of TARTs is unclear, but possibilities include adrenal rests, interstitial cells, and pluripotent ectopic adrenal cortex remnant cells of the testicular stroma stimulated by elevated androgen levels during embryogenesis which differentiate and grow under the influence of chronically elevated ACTH [4]. Multiple case reports and case series have found positive correlations between age, pubertal stage, and the presence of TARTs, with the highest prevalence in adults, lending support to the hypothesis that dysregulation of the follicular stimulating hormone (FSH)/luteinizing hormone (LH) axis may be an important factor in the development of TARTs [5–7]. The lesions are usually asymptomatic but can lead to structural damage, disorders in spermatogenesis, and infertility, and can grow in size to obliterate normal testicular parenchyma. It has been suggested by some authors that screening for TARTs during childhood in male patients with CAH due to 21-OHD is recommended given the potential implications involving fertility and the destruction of normal testicular parenchyma if inadequately treated [6]. Because of these potential complications, the importance of recognizing this clinical entity in men with CAH due to 21-OHD is of paramount importance for physicians.
The diagnosis of CAH due to 21-OHD is generally established by a combination of state-sponsored neonatal screening programs and clinical presentation. Ultrasonography is generally considered to be the first-line imaging modality for detection and characterization of TARTs given its widespread availability and significantly lower cost over other imaging modalities including computed tomography (CT) and magnetic resonance imaging (MRI). Imaging features on sonographic evaluation can show a wide variety of features, but tend to be sharply marginated and heterogeneously hypoechoic, with larger lesions containing calcifications or hyperechoic rims [7–9]. The diagnosis of TARTs after an initial work-up utilizing sonographic evaluation is usually made clinically. The general consensus among the interprofessional teams and scientific literature indicate that the great majority of individuals with TARTs will have bilateral testicular involvement, whereas other similar appearing entities including Leydig cell tumors will almost always have unilateral involvement [4].
The roles of CT and MRI in the evaluation of TARTs have not been adequately investigated. Because of the benign nature of TARTs, it is generally thought that further evaluation utilizing these modalities is not necessary in mild cases of TARTs when ultrasonography demonstrates no significant interval change over time [7]. It has been suggested that there are some intrinsic advantages to performing CT and/or MRI evaluation including formal evaluation of the enlarged adrenal glands, better image characterization across multiple sequences, and the evaluation of multiple small, discrete lesions versus larger, solitary lesions [7, 10]. If the extent of disease is being evaluated or if surgery is being considered, a higher imaging modality such as MR or CT would be preferable to ultrasound [10]. With regards to our patient, there were no further interval sequelae of his disease state that warranted further evaluation with a higher imaging modality.
Histologically, TARTs are similar in appearance and morphology to adrenocortical tissue, but may sometimes be mistaken for malignant Leydig cell tumors. Features which may be helpful on tissue evaluation to help discriminate between the two entities in addition to laterality include the presence of Reinke crystals (present in 25–40% of Leydig cell tumors, but absent in TARTs), and immunohistochemical stains such as synaptophysin (which shows strong reactivity to TARTs but no reactivity to Leydig cell tumors) [4, 11].
TARTs are typically treated with medications. It is generally accepted that the tumors often shrink in size with the administration of adequate amounts of exogenous glucocorticoids, as was seen in our case [12]. Dexamethasone, a long-acting glucocorticoid, is most effective in suppressing adrenocorticotropic hormone (ACTH), which is needed to help with regression in size of the TARTs. There is a balance between suppressing ACTH and over-treatment with glucocorticoids which may lead to iatrogenic Cushing syndrome. It is hypothesized that inadequate hormonal control may lead to the continued growth of these tumors, ultimately destroying normal testicular parenchyma and resulting in permanent infertility among other complications. However, TARTs may also be found in patients who were adequately treated, as was demonstrated in a recent case series [6]. Additionally, further studies have shown that some patients with poorly controlled CAH will never develop TARTs [13, 14]. This implies that inadequate hormonal control and medication non-compliance may be part of a continuum of factors in the development of TARTs. Nevertheless, hormone replacement therapy is advocated as the first-line treatment. Rarely, patients with chronic testicular pain refractory to hormone replacement therapy may be considered for further treatment including partial or total orchiectomy [10, 12]. In general, a normal semen analysis is the best evidence of good disease control without overtreatment, and sperm banking may be considered in young men who express a desire to preserve fertility.
In conclusion, it is important for both clinicians and radiologists to become familiar with finding and evaluating congenital anomalies in adults. Testicular imaging is of utmost importance in men with a history of CAH due to 21-OHD or to 11β-hydroxylase deficiency, and this case demonstrates the value of ultrasound in aiding the diagnosis of TARTs. The imaging findings may be suggestive of inadequate hormone replacement therapy. For the radiologist in particular, the recognition of this entity in the context of the clinical picture will help prevent unnecessary and life-altering treatments including orchiectomies. It is therefore crucial that once diagnosed, patients with TARTs adhere to medication and continue follow-up with physicians knowledgeable in the treatment of CAH.