Introduction
The level of hemoglobin (Hb) A2 (α2δ2) in a normal adult is about 2.0–3.2% of total Hb. An elevated Hb A2 between 4.0% and 8.0% is an important diagnostic marker for β-thalassemia carriers [1–3]. A study in a large cohort of β-thalassemia heterozygotes in Thailand detected Hb A2 corresponding to β-thalassemia mutations in the range of 5.0 ±0.6% to 7.0 ±0.8%. Interaction of β-thalassemia with α-thalassemia could alter the level of Hb A2 but this does not affect the diagnosis of the cases [4]. In some cases, the levels of Hb A2 are higher than normal but are not within the diagnostic range for a typical β-thalassemia carrier, making it difficult to make the diagnosis. This so-called borderline Hb A2 usually ranges between the levels found in normal and classic β-thalassemia carriers, which are 3.2–3.9% [5, 6]. Therefore, accurate diagnoses of these cases are usually made through molecular identification of the β-thalassemia mutations [7]. However, this is not practical in a large population screening in an area where thalassemia is common and molecular testing is not available. Borderline Hb A2 is prevalently reported in many populations. Several causes of reduced Hb A2 have been noted including reduced δ-globin chain synthesis in δ-thalassemia or δ-chain variant, reduced Hb A2 tetramer assembly in α-thalassemia, a silent β-thalassemia mutation, α-globin gene triplication and some acquired conditions such as iron deficiency, lead poisoning, sideroblastic anemia, and myeloproliferative disorders [8]. These causes could generally explain only a small fraction of the cases with borderline Hb A2 in the populations [2, 7–10]. This indicates the involvement of other genetic factors. Among these factors is an erythroid-specific transcription factor, Krüppel-like factor 1 (KLF1), which plays an important role in erythropoiesis and Hb switching. KLF1 mutations have been found to be associated with several hematological disorders, including hereditary persistence of fetal hemoglobin (HPFH), congenital dyserythropoietic anemia type IV, and transfusion-dependent hemolytic anemia. It has been found that KLF1 mutations could also ameliorate the clinical severity of β-thalassemia disease [11–13]. Association of KLF1 mutations with borderline Hb A2 have been noted in multiple populations including Italian, Sardinian, Chinese and Saudi [7, 9, 10, 14]. We have also reported the association of KLF1 mutations with higher phenotypic expression of Hb A2 and Hb F in Hb EE syndrome [15, 16]. In this study, we examined globin genes and KLF1 mutations to determine the molecular features underlying borderline Hb A2 in a large cohort of the Thai population.
Material and methods
Subjects and hematological analysis
Ethical approval of the study protocol was obtained from the Institutional Review Board (IRB) of Khon Kaen University, Thailand (HE562059). A retrospective review of laboratory findings was carried out on 21,657 Thai subjects encountered at our ongoing thalassemia screening program at the Centre for Research and Development of Medical Diagnostic Laboratories, Khon Kaen University, Thailand. A total of 202 subjects was selectively recruited based on Hb analysis with Hb A2A pattern and Hb A2 levels between 3.5 and 4.0%. Subjects with known heterozygous β-thalassemia and elevated Hb A2 (> 4.0%), thalassemia diseases or Hb variants and iron deficiency anemia were excluded. Routine hematological parameters were recorded using a standard blood cell counter. Hemoglobin analysis and quantification were performed using Hb high-performance liquid chromatography (Variant II analyzer, Bio-Rad Laboratories, Hercules, CA, USA) or capillary electrophoresis (Capillarys 2 Flex Piercing; Sebia, Lisses, France).
Molecular analysis
Genomic DNA was prepared from peripheral blood leukocytes using the standard method. Identifications of β-thalassemia mutations, α0-thalassemia (SEA & THAI deletions) and α+-thalassemia (3.7 and 4.2 kb deletions), Hb Constant Spring and Hb Paksé mutations are performed routinely in our laboratory using PCR and related methods described elsewhere [17, 18]. Screening for α-globin gene triplication (ααα haplotype) and δ-globin gene variants previously documented in Thailand was done using PCR assays as described previously [19, 20].
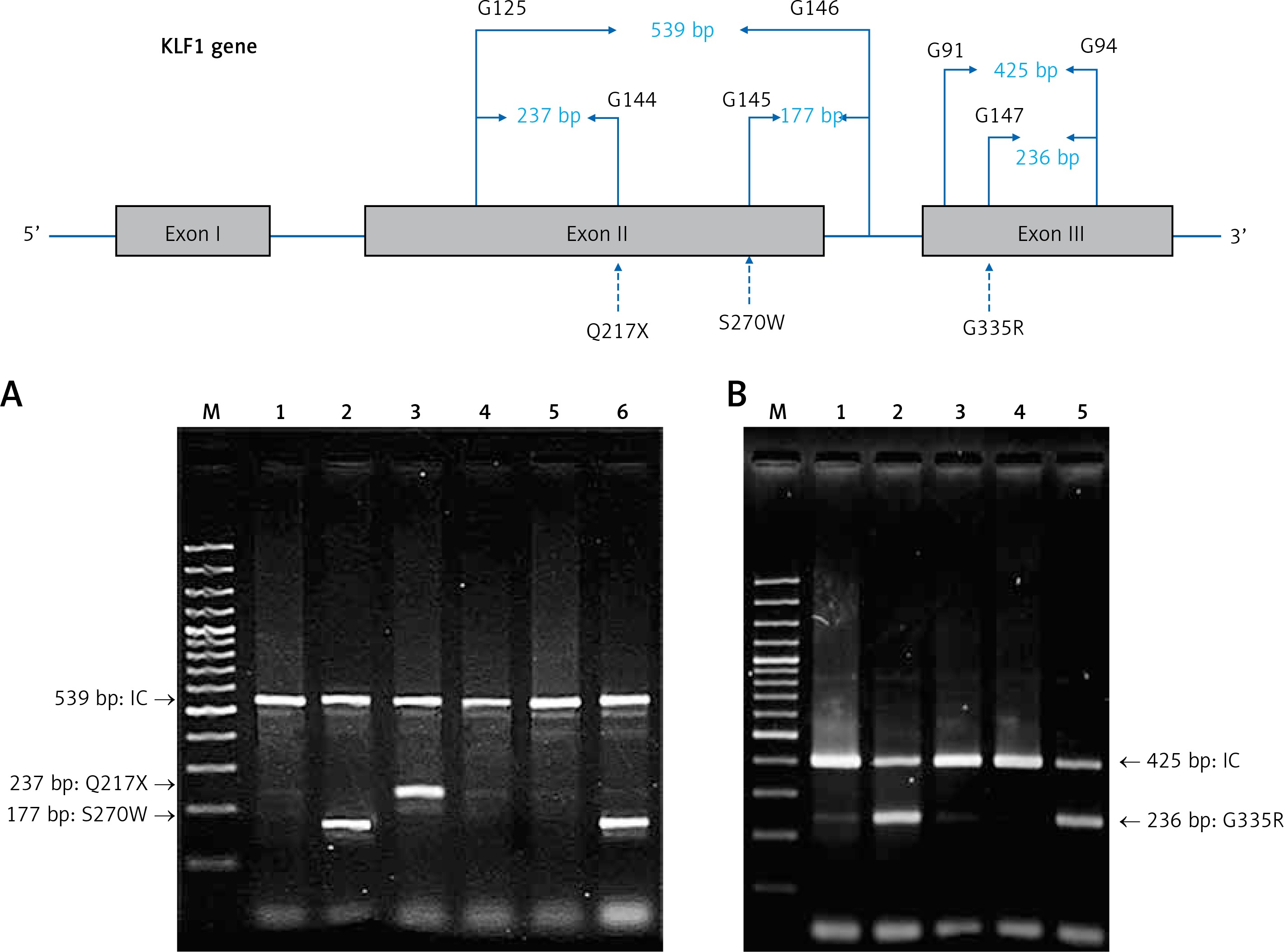
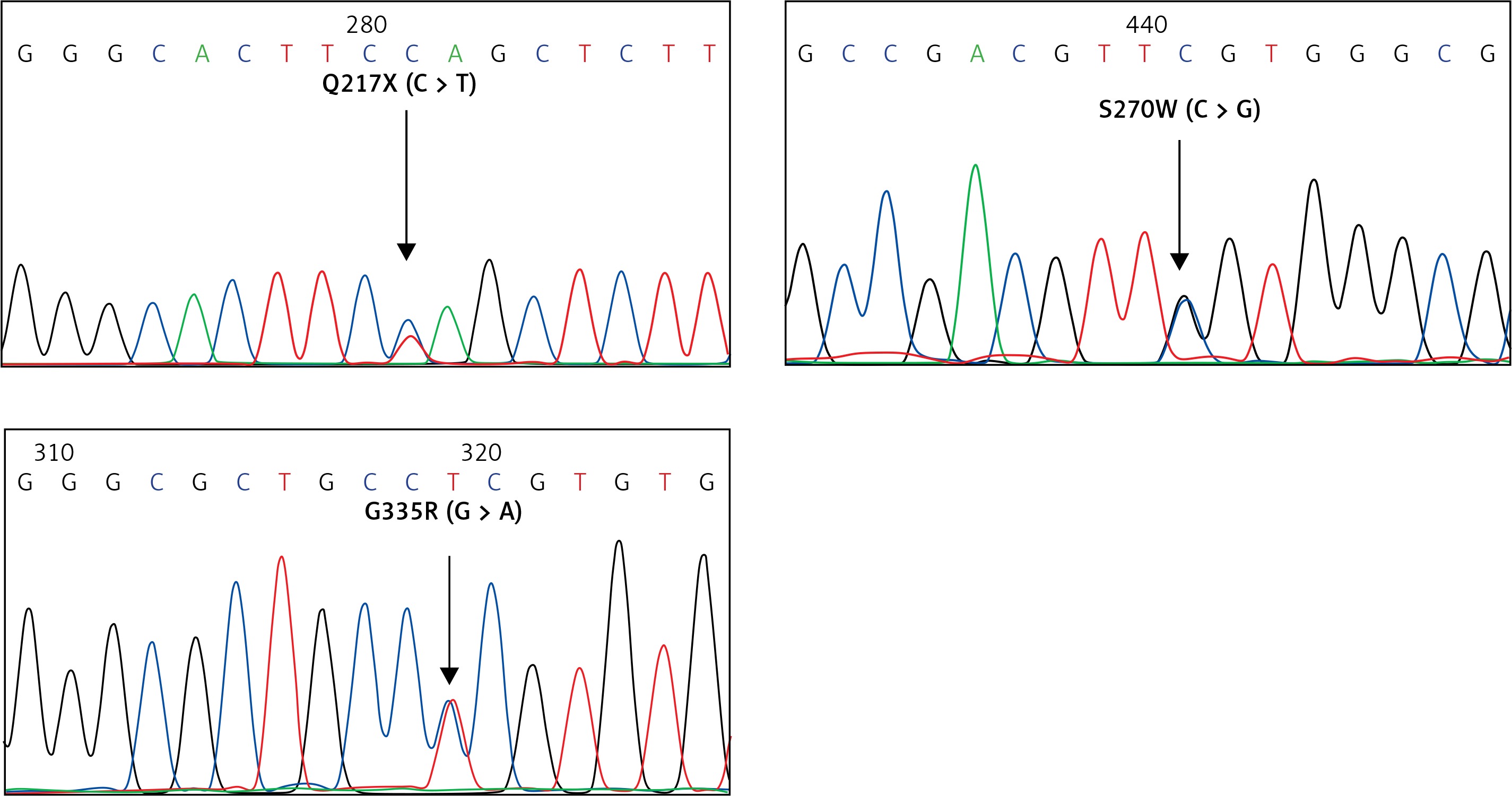
For the KLF1 gene, four common KLF1 mutations found previously in the Thai population – G176AfsX179, -154(C-T), R328H and T334R – were examined using allele-specific PCR as described [15, 16]. Other rarer mutations including Q217X, S270W and G335R were identified using allele-specific PCR assays developed in this study as shown in Figure 1. These mutations were then confirmed by DNA sequencing (Figure 2). The Q217X and S270W mutations were detected in a multiplex PCR manner. With this system, the Q217X and S270W specific primers, G144 (5′-GAGCCCGCGGAAGAGCTA-3′) and G145 (5′-CAAGCGAGGCCGACGTTG-3′), were used with primers G125 (5′-GCTCCCGACGCCTTCGT-3′) and G146 (5′-TGTAACTACAGCGGGCGCC-3′), respectively, to produce fragments of 237 bp and 177 bp specific for the two mutations. The 539 bp fragment was an internal control generated from primers G125 and G146. The PCR reaction (50 μl) contained 50 ng of DNA, 0.6 pmol of primers G144 and G146, 1.2 pmol of primer G125, 0.33 pmol of primer G145, 200 mM dNTPs, 100 mM Tris HCl pH 8.3, 50 mM KCl, 3 mM MgCl2, 5% DMSO, 1 M Betaine and 1 unit of Taq DNA polymerase (New England Biolabs Inc., MA, USA). PCR was performed with an initiation heating step at 94°C for 3 min followed by 35 cycles of 94°C for 30 s and 67°C for 1.30 min. In contrast, the G335R mutation was detected under a monoplex allele-specific PCR with the specific primer G147 (5′-GCCACTACCGGAAACACACGA-3′) and the common primer G94 (5′-TCCAGGAGAGGGTCCATTCGT-3′) which produces a 236 bp specific fragment. An internal control of 425 bp was generated from primers G91 (5′-CAGCCCAGGCTGAGTAAAGGG-3′) and G94. The reaction mixture (50 μl) contained 50 ng DNA, 0.72 pmol of primers G147, 0.6 pmol of primer G91 and G94, 200 mM dNTPs, 100 mM Tris HCl pH 8.3, 50 mM KCl, 3 mM MgCl2 and 1 unit of Taq DNA polymerase (New England Biolabs Inc., MA, USA). PCR was performed with an initiation heating step at 94°C for 3 min followed by 35 cycles of (94°C for 30 s and 69°C for 1.30 min). The amplification was performed using the TProfessional Standard Thermocycler (Biometra, GmbH, Goettingen, Germany). The amplified product was separated on 2% agarose gel electrophoresis, stained with ethidium bromide (0.5 μg/ml) and visualized under UV light, shown representatively in Figure 1.
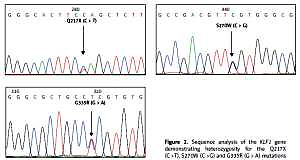
Figure 1
Identification of three KLF1 mutations, namely Q217X, S270W and G335R, by allele specific PCR assays. The mutations, locations and orientations of all primers used as well as the sizes of amplified fragments are depicted. In both representative gel electrophoresis, M is 100 bp DNA ladder. A – 1: normal control, 2 and 6: carriers of S270W mutation, 3: carrier of Q217X mutation, 4 and 5: negative for S270W and Q217X mutations. B – 1: normal control, 2 and 5: carriers of G335R mutation, 3 and 4: negative for G335R mutation
Statistical analysis
Mean and standard deviation were used to describe the hematological parameters of the subjects. The difference in the medians of MCV, Hb A2 and Hb F was analyzed using the Kruskal-Wallis test and the Mann-Whitney U test. A p-value of < 0.05 was considered statistically significant. Box plot and statistical analysis were done using Minitab version 14 (Minitab Inc., USA).
Results
Among 202 subjects with borderline Hb A2, DNA analysis revealed that 70 (34.7%) were positive for molecular defects in the α-globin gene (n = 48) and β-globin gene (n = 22) as shown in Table I. Forty eight subjects (23.8%) were found to carry KLF1 mutations including 30 pure heterozygotes, 8 compound heterozygotes and 10 double heterozygotes for KLF1 mutations and α-globin gene defects. No genetic interaction between KLF1 and β-thalassemia was observed. In the remaining 84 (41.5%) subjects, no molecular defects in the α- and β-globin genes or in the KLF1 gene were found.
Table I
Hematological parameters, KLF1 mutations and globin genotypes found in 202 Thai subjects with borderline Hb A2
| Mutations | n | % | Hb Typing | RBC [1012/l] | Hb [g/dl] | Hct [%] | MCV [fl] | MCH [pg] | MCHC [g/dl] | |
|---|---|---|---|---|---|---|---|---|---|---|
| Hb A2 (%) | Hb F (%) | |||||||||
| No mutation | 84 | 41.5 | 3.7 ±0.2 | 1.1 ±1.8 | 4.6 ±0.8 | 12.4 ±1.9 | 37.6 ±5.9 | 81.9 ±9.9 | 27.1 ±3.6 | 33.1 ±1.6 |
| Globin gene mutations: | 70 | 34.7 | ||||||||
| α-thalassemia carriers: | 48 | 23.8 | ||||||||
| -α3.7/αα | 34 | 16.9 | 3.6 ±0.4 | 0.4 ±0.4 | 4.9 ±0.8 | 12.4 ±1.6 | 37.8 ±5.1 | 77.9 ±4.7 | 25.6 ±1.7 | 32.6 ±1.3 |
| αCSα/αα | 12 | 5.9 | 3.7 ±0.2 | 1.3 ±1.6 | 4.8 ±0.6 | 12.1 ±1.9 | 38.2 ±5.4 | 79.2 ±4.7 | 25.2 ±2.0 | 31.7 ±1.1 |
| αPSα /αα | 2 | 1.0 | 4.0, 3.5 | 0, 0 | 4.6 | 11.4 | 34.2 | 70, 75.2 | 22, 25.1 | 33.3 |
| β+-thalassemia carriers*: | 22 | 10.9 | ||||||||
| β+-thal trait | 17 | 8.4 | 3.8 ±0.1 | 2.4 ±3.4 | 4.9 ±0.9 | 11.7 ±2.0 | 35.1 ±5.3 | 72.4 ±11.8 | 23.8 ±5.1 | 32.6 ±2.0 |
| β+-thal trait with -α.7/αα | 2 | 1.0 | 3.8, 3.7 | 0, 0.6 | na | na | na | 80.7 | 26.9 | na |
| β+-thal trait with αCSα/αα | 1 | 0.5 | 4.0 | 0.9 | 4.5 | 11.4 | 35.3 | 78 | 25.1 | 32.2 |
| β+-thal trait with --SEA/αα | 1 | 0.5 | 3.9 | na | 5.5 | 12.6 | 38 | 69.2 | 22.8 | 33 |
| β+-thal trait with --SEA/αCSα | 1 | 0.5 | 3.5 | 1.9 | 4.0 | 6.9 | 24.2 | 61 | 17.5 | 28.8 |
| KLF1 mutations: | 38 | 18.8 | ||||||||
| KLF1 trait | 30 | 14.8 | 3.7 ±0.2 | 2.3 ±2.1 | 5.0 ±0.7 | 13.3 ±1.5 | 40.8 ±5.1 | 80.9 ±8.2 | 26.5 ±2.5 | 32.5 ±1.6 |
| compound heterozygous KLF1 | 8 | 4.0 | 3.7 ±0.2 | 2.7 ±1.9 | 5.1 ±0.9 | 12.4 ±2.3 | 36.8 ±6.0 | 74.9 ±9.7 | 24.8 ±3.9 | 33.9 ±1.1 |
| KLF1 mutations with globin gene mutations: | 10 | 5.0 | ||||||||
| KLF1 trait with -α3.7/αα | 7 | 3.5 | 3.7 ±0.2 | 1.1 ±0.9 | 5.9 ±0.3 | 13.9 ±1.4 | 43.5 ±3.8 | 73.3 ±5.1 | 23.3 ±2.1 | 30.4 ±2.7 |
| KLF1 trait with αCSα/αα | 1 | 0.5 | 3.5 | 0 | na | na | na | na | na | na |
| KLF1 trait with --SEA/αα | 1 | 0.5 | 3.6 | 4.2 | 6.3 | 12.3 | 41 | 65 | 19.6 | 30.3 |
| Compound heterozygous KLF1 with -α3.7/αα | 1 | 0.5 | 3.6 | 0 | 5.4 | 12.6 | 38.4 | 71 | 23.4 | 34 |
As shown in Table I, among 48 subjects with α-thalassemia, 34 were heterozygous for α+-thalassemia with 3.7 kb deletion (-α3.7/αα), 12 were heterozygous for Hb Constant Spring (αCSα/αα) and 2 were Hb Paksé carriers (αPSα/αα). Interestingly, among 22 β-thalassemia carriers, only mild β+-thalassemia mutations were identified including Hb Malay or CD19 (HBB: c.59 A > G) in 10 cases, NT -31 (HBB: c.-81 A > G) in 5 cases, Hb Dhonburi or CD126 (HBB: c.380 T > G) in 3 cases, NT -28 (HBB: c.-78 A > G) in 3 cases and NT -50 (HBB: c.-100 G > A) in 1 case. Double heterozygosity for β+-thalassemia and α+-thalassemia (-α3.7/αα), Hb Constant Spring (αCSα/αα), α0-thalassemia (--SEA/αα) and Hb H-CS disease (--SEA/αCSα) were identified in two, one, one and one subjects, respectively. Thirty-eight of 202 (18.8%) subjects had the KLF1 mutations alone including 30 pure heterozygotes and 8 compound heterozygotes. Double KLF1 and α-thalassemia mutations were identified in 10 (5.0%) subjects including 7 with -α3.7/αα, 1 with αα/αCSα, 1 with α0-thalassemia with SEA deletion (--SEA/αCSα) and 1 compound heterozygous for two KLF1 mutations and α+-thalassemia (-α3.7/αα).
Table II summarizes the hematological findings of 48 subjects with KLF1 mutations according to α-globin genotypes. We detected 7 different KLF1 mutations among these 48 subjects. The most common KLF1 mutation found in 17 of the 48 (35.4%) subjects was the G176AfsX179 frameshift mutation. Four missense mutations – T334R, S270W, R328H and G335R – were found in 7 (14.6%), 6 (12.5%), 4 (8.3%) and 2 (4.2%) cases, respectively. The remaining mutations detected were a nonsense mutation Q217X (n = 1; 2.1%) and the -154 C>T promoter mutation (n = 2; 4.2%). Nine subjects were found to be compound heterozygotes for two different KLF1 mutations: -154 (C>T)/ Q217X (n = 6), G176AfsX179/ S270W (n = 2) and T334R/ R328H (n = 1). As expected, most of the subjects with α-thalassemia were found to have lower MCV & MCH values as compared to those without α-thalassemia. However, it is apparent that co-inheritance of KLF1 mutation does not worsen hematological features of these α-thalassemia patients.
Table II
Hematologic parameters of 48 subjects with KLF1 mutations and different α-globin genotypes
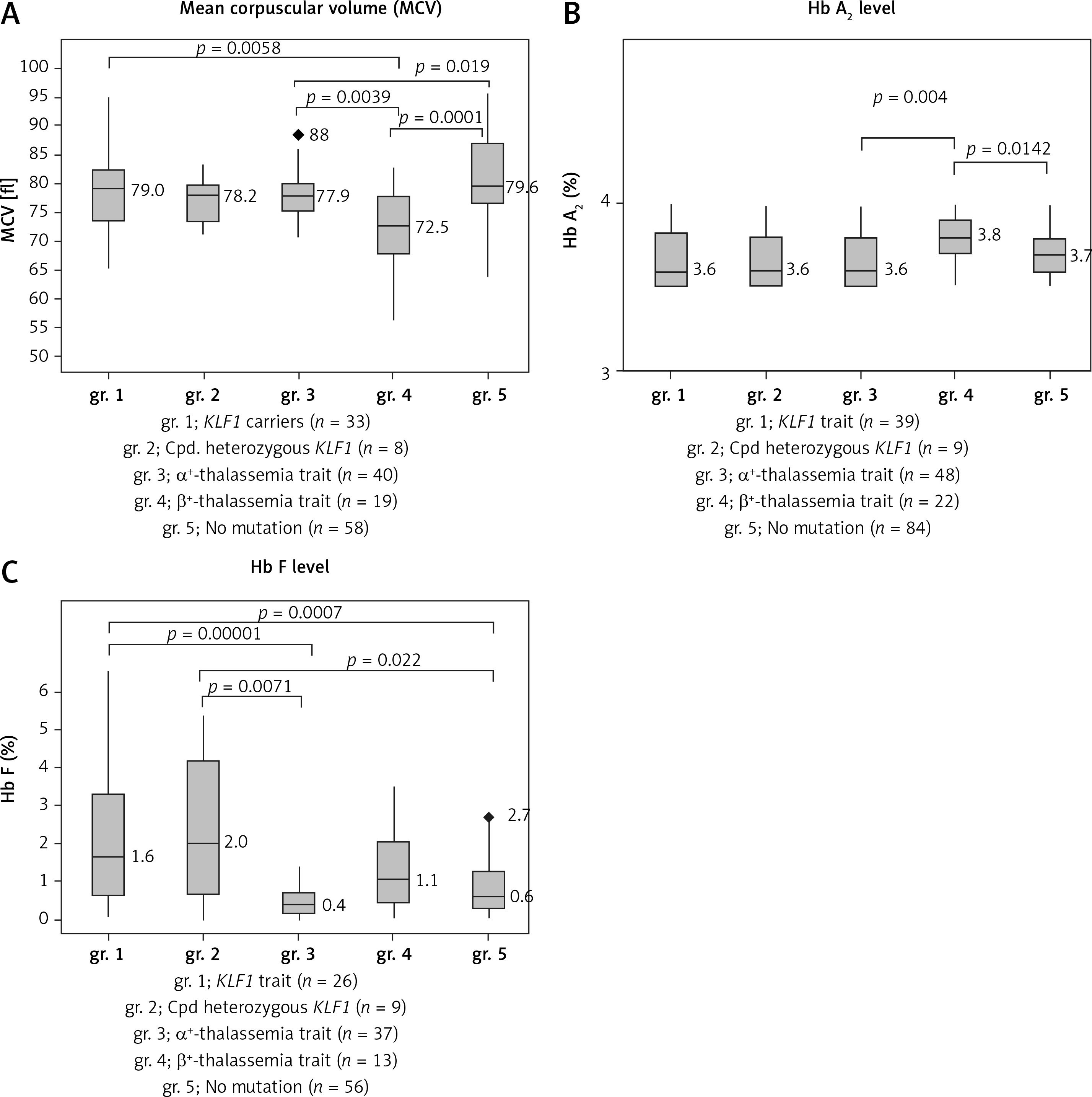
In order to visualize the effect of KLF1 mutations on the levels of MCV, Hb A2 and Hb F, box plots of MCV, Hb A2 and Hb F were presented comparatively in Figure 3 for gr. 1, KLF1 carriers; gr. 2, compound heterozygotes for KLF1 mutations; gr. 3, α+-thalassemia trait; gr. 4, β+-thalassemia trait and gr. 5, subjects with no mutation. Significantly, the lowest MCV value was noted for gr. 4, β+-thalassemia trait (Figure 3 A). It is also noteworthy in Figure 3 B that although all subjects had similarly borderline Hb A2 due to the selection criteria, a significantly higher level of Hb A2 was again observed in gr. 4, β+-thalassemia trait (p-value < 0.05 with the Kruskal-Wallis and the Mann-Whitney U tests). For Hb F, subjects with KLF1 mutations in gr. 1 and gr. 2 had significantly higher levels as compared to those of the remaining three groups without KLF1 mutations (Figure 3 C).
Figure 3
Box plots showing the distributions of MCV (A) Hb A2 (B) and Hb F (C) in five groups of Thai subjects with borderline Hb A2 as indicated along with number of cases with data available (gr. 1–gr. 5). Statistical comparison was done using the Kruskal-Wallis test and the Mann-Whitney U test and significant p-values between two comparable groups are indicated
Discussion
Elevated Hb A2 is an important marker for diagnosis of β-thalassemia carrier. In countries with a high prevalence of thalassemia, individuals with normal or elevated levels of Hb A2 are generally not problematic in routine diagnostics. However, those with borderline Hb A2 are difficult to diagnose. Previous studies in multiple populations have demonstrated different molecular defects in α-, β- and δ-globin genes as well as in the KLF1 gene [5, 7–10]. However, the results of these studies cannot be compared directly because of the differences in the Hb analyzers used in quantification of Hb A2, the molecular basis of β-thalassemia in the population, and the definitions of borderline Hb A2. For example, the definitions of borderline Hb A2 were 3.1–3.9% in the studies of Italian [5] and Saudi [10] subjects, 3.3–4.1% in the Sardinian population [9] and 3.3–4.0% in the Chinese [7] population. In Thailand, the value of Hb A2 > 4.0% has been used for diagnosis of β-thalassemia carriers [21]. Recently, Thailand’s Ministry of Public Health changed this cut-off value to > 3.5% in the Clinical Practice Guideline (CPG) for laboratory diagnosis of thalassemia. We have noted in our ongoing thalassemia screening program that this change has increased the number of referral cases for molecular analysis of β-thalassemia and a large number of cases with no β-thalassemia mutation. This so-called false positive has rendered genetic counseling difficult and affected the prevention and control program in the country. Since no information on the molecular characteristics of borderline Hb A2 in the Thai population is known, we looked at this matter in this study.
The level of Hb A2 between 3.5 and 4.0% was defined as borderline Hb A2 in this study and a total of 202 subjects were recruited. We found that approximately 62.0% of the cases had MCV < 80 fl and 78.8% had MCH < 27 pg (Table I), which are quite similar to those of β-thalassemia carriers identified in the region [4]. However, as many as 84 out of 202 (41.5%) subjects were found to carry no mutations in α-, β- and δ-globin genes and the KLF1 gene. When compared to other groups with mutations, these 84 subjects had higher MCV (81.9 ±9.9 fl) and MCH (27.1 ±3.6 pg) values. It is therefore recommended to consider cases with borderline Hb A2 in combination with red blood cell indices before referring such cases for further molecular testing of thalassemia. This should lead to a significant reduction in the number of referral cases and workload of molecular testing.
Thalassemia mutations were found in 70 out of 202 (34.7%) subjects including 48 subjects with mild α+-thalassemia and 22 carriers of mild β+-thalassemia. Both are not the prime targets of thalassemia screening in Thailand, which focuses mainly on α0-thalassemia, β0-thalassemia, and Hb E [17, 21]. This finding is also the case for the Italian, Chinese and Sardinian studies, in which most of the β-thalassemia mutations identified are β+-thalassemia [5, 7, 9]. In our study, neither α-globin gene triplication nor a δ-globin gene defect was detected. All these 70 cases could be considered as false positives in thalassemia screening. This should have further implications for the prevention and control program since it is not necessary to do mutation analysis for this group of subjects. This should also lead to a reduction in the number of referral cases for molecular study. However, we still recommend doing a molecular investigation in cases whose partners are β0-thalassemia or α0-thalassemia carriers in order to evaluate the risk of having fetuses with severe β-thalassemia major associated with compound β0/β+-thalassemia and Hb H disease caused by the interaction of α0- and α+-thalassemias. It is also noteworthy, as shown in Figures 3 A and B, that subjects with β+-thalassemia had significantly lower MCV (p = 0.0001) and higher Hb A2 (p = 0.0142) values as compared to those of subjects with no mutations. This characteristic should help in recognizing cases that require further molecular investigation.
Of interest is the finding of a high prevalence of KLF1 mutations (48 of 202; 23.8%) in this group of Thai subjects with borderline Hb A2. As many as 7 mutations, including both pure and compound heterozygotes, were encountered (Table II). We noted that this prevalence is higher than those described in the Chinese population [7] and in Thai subjects with Hb E disorder [15, 16]. This might indirectly support the association of KLF1 mutation with a borderline Hb A2 phenotype in the Thai population. We have shown previously in Hb EE disease that subjects with KLF1 mutations had significantly higher Hb A2 and Hb F as compared to those without KLF1 mutations [15, 16]. An expression study also demonstrated an increased ratio of δ/δ+β expression in KLF1 mutation carriers and an increase in Hb A2 level [14]. As for the Chinese population, the most common KLF1 mutation observed in Thailand is G176AfsX179 [8]. This mutation has also been found to be associated with increased Hb F in many populations including Korean, Vietnamese and Chinese [22, 23]. This has now been confirmed in the Thai population as shown in Figure 3 C, i.e. those with KLF1 mutations had significantly higher Hb F as compared to other groups. They also had higher MCV levels and lower Hb A2 values as compared to those of β+-thalassemia carriers (Figures 3 A, B). However, we did not observe much difference in hematological parameters among heterozygous and compound heterozygotes for different KLF1 mutations in Thai subjects (Table II). None of them had any clinical symptoms either. The result indicated a broad spectrum of KLF1 variants [24–26]. However, as reported previously [27], we did observe more hypochromic microcytosis in individuals with concomitant inheritance of KLF1 and α-thalassemia mutations (MCV 73.0 ±4.9 fl, MCH 23.3 ±1.9 pg) as compared to those with KLF1 mutations alone (MCV 77.9 ±4.7 fl, MCH 25.4 ±1.8 pg).
In conclusion, we demonstrated for the first time the molecular characteristics of borderline Hb A2 in the Thai population in about half of the cases. Only a small proportion of them are β+-thalassemia carriers. Other genetic and non-genetic factors may be involved with borderline Hb A2 phenotype in the remaining subjects such as HBS1L-MYB and BCL11A variants [28], HIV-infected patients treated with zidovudine [29], and the coefficient of variation of the method used to measure Hb A2 [1, 30]. Nonetheless, our study confirmed that most of the Thai individuals with borderline Hb A2 would not be at risk of having fetuses with the three severe thalassemia syndromes targeted in a prevention and control program. Screening for multiple causative mutations corresponding to borderline Hb A2 in the general population may not be cost-effective. However, careful evaluation of these individuals using combined Hb A2, Hb F, and MCV values should help in effective selection of a small number of cases for further molecular study. This should lead to a significant reduction of workload for molecular testing and facilitate a prevention and control program for thalassemia in regions such as Thailand where thalassemia and hemoglobinopathies are exceptionally prevalent and heterogeneous.