
Introduction
Sjögren’s syndrome (SS) is a chronic, systemic, autoimmune disease characterized by inflammation and damage of exocrine glands, particularly salivary and lacrimal glands. The main clinical manifestation of SS is dry mouth (xerostomia) and dry eyes (xerophthalmia). This autoimmune disease may be primary (pSS); however, it is more commonly may associate other autoimmune rheumatic diseases as a secondary Sjögren’s syndrome (sSS).
The current pSS classification criteria 2016 ACR/EULAR contain the following conditions: confirmation of eye dryness by Schirmer test or/and score of ocular staining with lissamine green and fluoresceine (also Bijsterveld’s method of scoring is accepted), presence of inflammatory lymphocytic infiltrates with a focus score of ≥ 1/4 mm2 in labial salivary glands and anti-Ro/SSA antibodies positivity [1]. Moreover, 30% of affected individuals present with polyclonal hypergammaglobulinemia, and have rheumatoid factor [2]. Less common findings (in 20–30% of pSS patients) include detectable anticardiolipin antibodies, cryoglobulins, and low levels of complement components C3 and C4 [3].
The etiopathogenesis of pSS remains unclear, despite a number of research papers suggesting genetic, environmental, and hormonal factors potentially affecting epithelial function, autoantibody and cytokine formation, T-cell and B-cell activation, yet failing to offer a consistent presentation of the pathological process [4–7].
The characteristic anti-SSA/Ro52 autoantibodies, which are common in pSS, suggest the possibility of complement activation via the classical pathway by immune complexes (IC) formation and subsequent tissue damage. Moreover, the decrease in serum C3 and C4 levels detected in over 20% of pSS patients has not yet been explained, although it indicates the role of innate immunity in pSS pathogenesis.
Complement
The key functions of the complement system (CS) are protection against infections, removal of ICs and potentially harmful self-molecules, and modeling of the interaction between the innate and adaptive immunity [8, 9]. The CS comprises approximately 60 mutually cooperating effector proteins, regulatory proteins, and receptors. Like many other biological systems, the CS is governed by two mechanisms: amplification and self-inhibition. Protein cascade activation leads to the formation of membrane-attack complex (MAC), which causes cell membrane perforation. The process of self-inhibition involves quenching this complement amplification by regulatory proteins to protect the body’s cells from damage.
The CS can become activated via three pathways: classical, lectin, and alternative (Fig. 1). The classical pathway is induced by ICs, some viruses and bacteria, pentraxins, and cell fragments. Complement cascade activation via the lectin pathway is initiated by the binding of a mannose-binding protein to mannose residues on the pathogen surface. Lastly, the alternative pathway is activated spontaneously and continually; it predominates quantitatively over the classical pathway, and – uncontrolled – it could pose a danger to the body’s cells [8, 9].
Fig. 1
The three main activation pathways of complement: the classical, mannose-binding lectin, and alternative pathways (according to Bora NS [10] – modified). The proteins under examinations in red borders.
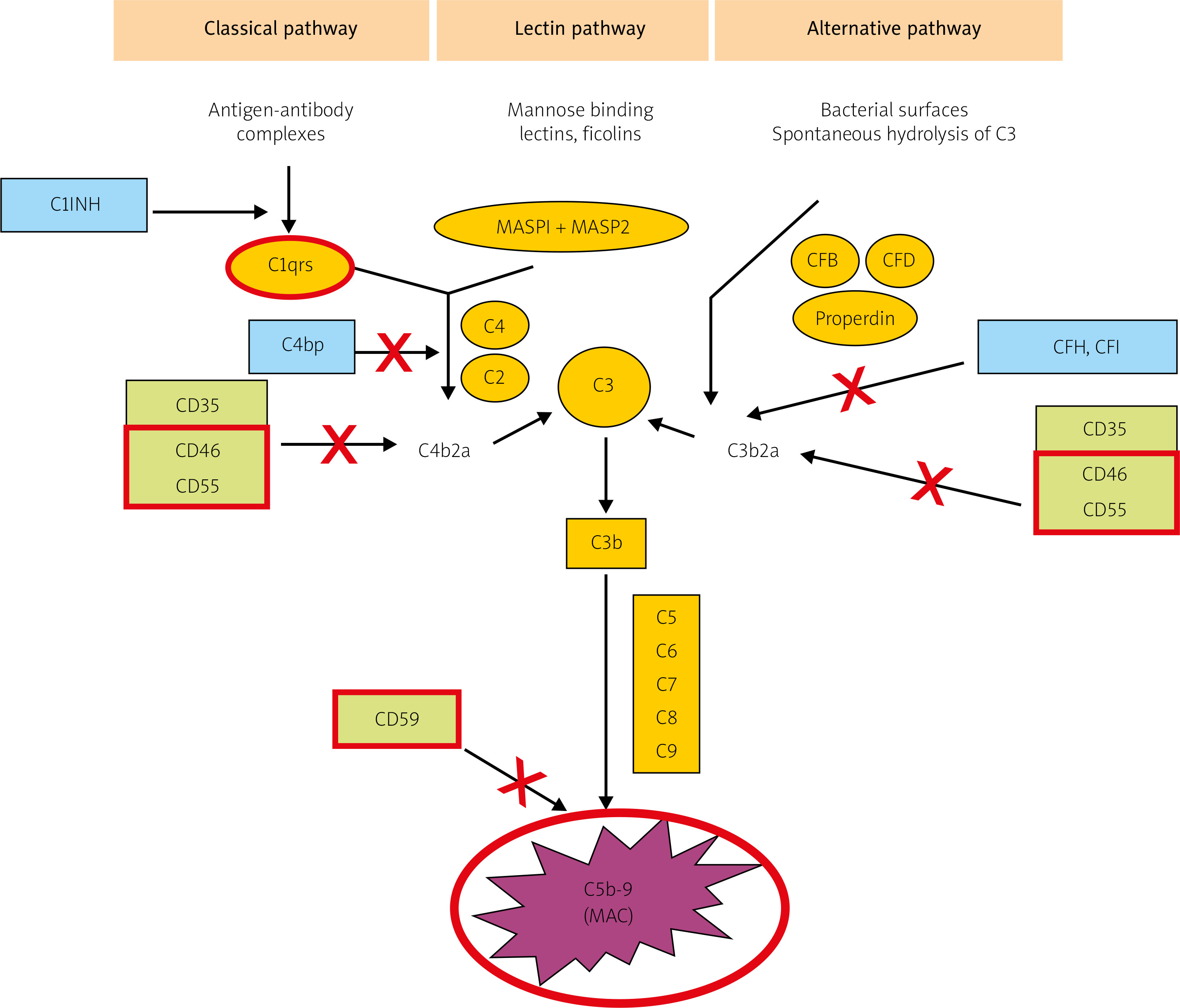
All three pathways converge at the stage of C3 activation. It is the C3-convertase and its derivatives (compounds containing C3b) that are the end-products of CS activation by most of its regulatory proteins. Membrane-bound representatives of these proteins include the decay-accelerating factor (DAF, CD55) and cofactors for the protease activity of complement factor I: membrane cofactor protein (MCP, CD46) and complement receptor 1 (CR1) [10, 11]. Soluble complement inhibitors that interact with C3 and its derivatives are factor H (for the alternative pathway) and factor I (for the classical pathway) (Fig. 1). Furthermore, protectin (CD59), anchored in the membranes of all blood cells, inhibits C9 polymerization, which protects the cell membrane from perforation. The soluble inhibitors vitronectin and clusterin exert similar effects.
There have been reports on complement regulatory protein dysfunction as an important element in the pathophysiology of autoimmune conditions [12–14].
Our search through the available literature yielded only two, partially relevant, reports on the expression of complement components and regulators in the labial salivary glands of patients with pSS [15, 16], despite the fact that over 20% of those patients have low C3 and C4 levels. Further, more detailed studies on the expression of complement cascade and regulatory proteins in labial salivary glands may become the next step toward elucidating the pathogenesis of pSS, which could be useful in developing new therapies.
The aim of presented study was to assess the expression of cascade proteins in labial salivary glands such as: C1q (a marker of classical complement activation), MAC (an indicator of the complement effector function, regardless of the activation pathway), and the membrane-bound complement regulatory proteins: MCP (CD46), protectin (CD59), and DAF (CD55).
Material and methods
Our study was conducted on tissue specimens archived at the Pathology Department of National Institute of Geriatrics, Rheumatology and Rehabilitation in Warsaw between 2015 and 2017. These specimens were paraffin blocks containing labial salivary gland biopsy samples collected from three patient groups (Table I):
Table I
Evaluated groups
| Characteristics | n | F | M | Age |
|---|---|---|---|---|
| Primary Sjögren’s syndrome | 20 | 18 | 2 | 52 (20–70) |
| Non-specific sialadenitis | 5 | 5 | 0 | 57 (41–74) |
| Normal salivary glands | 5 | 4 | 1 | 37 (17–49) |
Patients diagnosed with pSS based on the clinical presentation (following referral to the Pathology Department) and meeting the criterion of a focus score of ≥ 1 (n = 20).
Affected control group: with chronic non-specific sialadenitis (n = 5).
Control group: patients with normal salivary glands microscopic structure with no evidence of inflammation (n = 5).
Hematoxylin and eosin (HE)-stained specimens were examined for lymphocytic inflammatory infiltrates (focus score), secondary lymphoid follicles containing germinal centers (GC), and adenoid-tissue damage. Each sample underwent immunohistochemical (IHC) staining for C1q (a marker of classical complement activation), MAC (an indicator of the complement effector function, regardless of the activation pathway), and the membrane-bound complement regulatory proteins MCP, protectin, and DAF.
Paraffin sections (4 µm-thick) were deparaffinized in a standard way. Antigens were retrieved with Target Retrieval Solution, Low or High pH (Dako) according to the manufacturer’s instructions. Endogenous peroxidase activity was blocked with H2O2. Abcam antibodies were diluted with Dako Antibody Diluent with Background Reducing Components to obtain optimal concentrations: CD46 1 : 200, CD55 1 : 200, C5b-9 1 : 1000, CD59 1 : 100, C1q 1 : 200. Slides were incubated with primary antibodies in a humidified chamber for 30 minutes at room temperature. Dako REALTM EnVisionTM HRP Rabbit/Mouse and DAB (Dako) were used for visualization. Cell nuclei were stained with Harris’s hematoxylin. Sections were dehydrated and mounted with Histokitt (Carl Roth). Optimal antibody concentrations were determined on archived paraffin sections of lymph nodes and synovial membranes from rheumatoid arthritis patients.
These tissues also served as positive controls for IHC reactions. The procedures were performed without antibodies as negative controls – incubation with only Dako Antibody Diluent with Background Reducing Components.
Light microscopy was used to assess the location and classify the intensity of IHC reactions as negative or trace (–), slightly positive (+), moderately positive (++), or highly positive (+++). The expression of individual evaluated cell-membrane proteins was assessed in acinar (secretory) and ductal epithelial cells of salivary glands and in inflammatory cells. IHC staining for CD59 was done in such a way as to assess whether this molecule was expressed on the entire surfaces of acinar and ductal cells or on their luminal surfaces only. IHC reactions were also evaluated on endothelial surfaces and within vascular lumina (plasma).
The approval of the local ethics committee at the National Institute of Geriatrics, Rheumatology and Rehabilitation, Warsaw, Poland was obtained (decision 30.01.2020).
Results
Salivary gland biopsy specimens from patients diagnosed with pSS yielded focus scores ranging from 1 to 5. Glandular structure damage was observed in 11 out of the 20 evaluated cases, whereas 3 specimens contained secondary lymphoid follicles (Table II). HE staining in the 5 evaluated cases of chronic non-specific sialadenitis (affected controls) showed dispersed lymphocytes. The 5 non-affected controls showed unremarkable microscopic structure of the salivary glands, with no inflammatory infiltrates.
Table II
Results of IHC reactions in salivary glands in pSS
All cases of anti-C1q IHC staining of gland structures in the three study groups were negative (Tables II, III, IV), with only slightly positive (+) reactions observed in the plasma (in some vascular lumina), which proved the correct execution of staining protocols.
Table III
Results of IHC reactions in non-specific sialadenitis
Table IV
Results of IHC reaction in normal salivary glands
Likewise, no MAC (C5b-9) expression was detected in gland tissue from either pSS or non-specific sialadenitis patients, with only slightly positive reactions observed in the plasma (Tables II, III). Normal salivary glands stained with anti-C5b-9 showed negative reactions (Table IV).
Anti-MCP (CD46) IHC staining produced varied reactions. Fifty percent of the salivary glands with a focus score of ≥ 1 showed no MCP expression either on acinar or ductal cell membranes. The remaining glands showed a slightly positive (+) or inconsistent reaction (10–20% of acinar cells and few ducts). More intense reactions (from + to +++) were observed on the cell membranes of some infiltrate-forming lymphocytes (Fig. 2A). There were no differences between biopsy samples with and without glandular tissue damage or those with or without secondary lymphoid follicles.
Fig. 2
IHC staining for CD46 (A–C). pSS – intensive reaction of lymphocytes – arrows (A), non-specific sialadenitis: markedly positive (+++) reactions on the surface of ductal epithelium (B), normal salivary glands: homogeneously positive reaction (+++) with anti-MCP on nearly all acinar cell membranes (C). IHC staining for CD59 (D–F). pSS – CD59 homogeneously pronounced positive expression only on luminal surface of acinar and ductal cells (D), non-specific sialadenitis: luminal-surface reactions (+)/(++), with positive (+)/(++) reactions observed on the entire surface of acinar cells (E), normal salivary glands: predominantly highly positive reactions on the entire surfaces of acinar and ductal epithelial cells (F). All pictures magnification × 200.

Non-specific sialadenitis specimens showed markedly positive (++/+++) reactions with anti-MCP antibodies on the surface of ductal epithelium (Fig. 2B). Acinar and inflammatory cell membranes produced focal, less pronounced (+) reactions.
Non-affected control salivary gland specimens produced a homogeneously pronounced positive reaction (+++) with anti-MCP on nearly all acinar and ductal cell membranes (Fig. 2C).
Protectin (CD59) expression in pSS glands varied in intensity from (+) to (+++) and was present only on the luminal surface of acinar and ductal cells (Fig. 2D). Non-specific sialadenitis specimens showed less intense luminal-surface reactions (+)/(++), with positive (+)/(++) reactions observed on the entire surface of acinar cells (Fig. 2E). Non-affected control salivary glands showed inconsistent IHC reactions; with predominantly highly positive reactions on the entire surfaces of acinar and ductal epithelial cells (Fig. 2F).
Slightly positive IHC reactions with anti-DAF (CD55) were present in some pSS and non-specific sialadenitis specimens, though only on paraglandular connective-tissue fibers. Normal salivary glands showed no DAF expression.
Discussion
Labial salivary glands constitute adequate specimens for the assessment of the type and intensity (focus score) of chronic inflammation in the diagnostics and monitoring of pSS. Inflammatory cell infiltrates may disturb glandular function in several mechanisms (by tissue damaging cytotoxic cells, the secretion of cytokines that activate interferon-associated pathways, or local autoantibody production) [5]. Our study on the expression of bioactive molecules (i.e. complement components and regulatory proteins) may be an important step towards elucidating these mechanisms.
There have been a relatively large number of reports on low serum C3 and C4 levels in pSS patients and the predictive value of this finding for lymphoma development [2, 3]. However, the distribution of complement components and regulators in salivary glands has been addressed scantily and incompletely [15, 16].
Our study showed no expression of either C1q or MAC in the labial salivary glands classified as affected by SS. This suggests that the autoimmune process of pSS involves no local complement activation via the classical (no C1q expression), alternative, or lectin (no MAC expression) pathways. The lack of local classical-pathway activation – despite evidence of peripheral complement component depletion (low serum C3 and C4 levels) – may indicate that the formation of ICs in salivary glands is not an important part of the inflammatory process and the observed tissue damage results from a different mechanism than MAC-induced cytolysis. Interestingly, a Chinese study [16] demonstrated positive C1q expression via IHC staining in labial salivary glands of 71 patients with pSS. The discrepancy between those and our findings may be due to their use of Zhongjin Jinqiao Biotechnology Co. (Beijing China) rabbit polyclonal antibodies and our use of Abcam mouse anti-C1q monoclonal antibodies.
The expression of complement regulatory proteins MCP (CD46), DAF (CD55), protectin (CD59) on acinar cell membranes in normal salivary glands was already demonstrated in the early 1990s [17–19].
Membrane-attack complex (CD46) is a transmembrane protein present on all nucleated cells. Its main role as a cofactor for factor I in C3b and C4b cleavage (which halts component-cascade activation) is to protect autologous cells from damage by MAC. MCP is also a T-cell co-stimulator, inducing T-cell (CD4+) differentiation into Treg, which secrete anti-inflammatory cytokines (e.g. IL-10) and thus suppress immune response and prevent autoimmunization [20, 21].
IHC staining of salivary glands showed no expression of CD46 in acinar or ductal cells in 50% of pSS patients, with slight (+) or inconsistent responses in the remaining half. Conversely, lymphocytes showed locally high expression of CD46 – from (+) to (+++) – particularly on the periphery of inflammatory foci. Study results reported by Cuida at al. [15] were somewhat different, with MCP expression observed on the luminal surfaces of acinar and ductal epithelium. However, the study sample size should not be overlooked, with 20 cases evaluated in our study and 5 in cited study. Nonetheless, our findings are consistent with those reported by authors regarding CD46 expression on inflammatory cell membranes in pSS. This leaves room for further research to determine the phenotype of inflammatory cells expressing CD46, which might show whether MCP expression indeed correlates with T-cells obtaining the Treg anti-inflammatory phenotype.
It is difficult to explain the highly positive IHC reaction with anti-CD46 in non-specific sialadenitis being limited to ductal epithelial cells (Fig. 2B), with a less pronounced reaction observed on acinar and inflammatory cells. Likewise inexplicable is the fact that CD46 expression in normal salivary glands was different, with a homogeneous, highly positive reaction on acinar and ductal epithelial cells (Fig. 2C). Is it only such “expansive” CD46 expression that protects salivary gland cells from autoimmunity-induced destruction? Questions also arise about possible complement modulators. Answering these questions calls for employing a different study methodology (evaluating gene transcription and protein incorporation into cell membranes).
Protectin, or membrane inhibitor of reactive lysis (MIRL, CD59), is a glycosylphosphatidylinositol (GPI) – anchored membrane-bound glycoprotein, which is a structural component of peripheral blood cells (leukocytes and erythrocytes). Protectin is expressed on the endothelial surface and epithelia of exocrine-glands (including salivary glands) [18]. It inhibits the formation of MAC (C5b-9) by binding to C8 and C9 and preventing the incorporation of multiple C9 molecules into MAC. Moreover, protectin plays a role in signal transduction for T-cell activation and thus modulates cell-mediated immunity [22–25]. Another, recently suggested role of CD59 was that its activation on target-cell membranes may potentiate NK-cell-mediated cytotoxicity [24]. Additionally, CD59 expressed on NK-cells may enhance their cytolytic activity [25].
Similar to the Cuida et al. research [15], our study showed CD59 expression on luminal surfaces of acinar and ductal epithelial cells in pSS (Fig. 2D). We observed no differences in CD59 expression in salivary glands with and without microscopic evidence of glandular tissue damage. Therefore, this finding provided no explanation as to how the presence of CD59 on gland cells may affect the extent of their damage. Nonetheless, the noticeable differences in CD59 expression in the salivary glands in pSS and those in non-specific sialadenitis – with weaker IHC reaction intensity in the latter (Fig. 2E) – demonstrated a dissimilar character of inflammatory processes in these two conditions. IHC staining showed no protectin on inflammatory-cell membranes.
One interesting finding was that normal salivary glands showed marked (++/+++) CD59 expression all over acinar cells, not only on their luminal surface (Fig. 2F). This distinction was not shown in Cuida et al. [15] work. Although CD59 expression have been long shown to be affected by cytokines [14, 26], its regulatory mechanisms are not fully understood. CD59 may reach the cell membrane via exocytosis [27] and be subsequently freed from its GPI bond via specific phospholipase C or proteolytic enzymes [28]. Possibly, during the course of sialadenitis, tissue cytokines alter CD59 expression on acinar cell membrane, whereupon it is left only on the luminal surface (which is not in direct contact with surrounding tissues). This theory requires further studies.
Interestingly, in rheumatoid arthritis (RA) CD59 expression was shown to be absent on synovial cells and reduced on interstitial cells and the endothelial cells of synovial membrane blood vessels [14]. Moreover, studies on an antigen-induced arthritis (AIA) model in CD59-deficient mice demonstrated an increased number of MACs in the synovial membrane – and, consequently, greater tissue damage compared with the control group – with subsequent improvement following CD59-deficiency correction [29]. It turns out that one of the mechanisms of the anti-inflammatory effect of statins observed in RA mice may be an increased CD59 expression in tissues, which results in reduced complement activation [30]. A similar anti-inflammatory effect was observed in about a dozen clinical trials in RA patients [31].
Decay-accelerating factor (CD55) is a globular glycoprotein anchored to the cell membranes of blood cells, cells in a direct contact with plasma, and epithelial cells lining body cavities [19]. Decay-accelerating factor inhibits the formation of C3 and C5 convertases, thus limiting complement cascade activation [32]. It also acts as a modulator reducing the T-cell-mediated response [33]. As a CD97 ligand, DAF (together with CD97) induces T-cell and B-cell proliferation [32].
Surprisingly, our study showed no CD55 expression either in normal salivary glands or those affected by non-specific sialadenitis or pSS. Trace CD55 expression was only detected in interstitial salivary gland tissue, which was consistent with previous Cuida’s et al. [15] findings. In models of various inflammatory diseases, including autoimmune ones, the lack of CD55 has been associated with a greater severity of clinical manifestations [34]. However, Hoek et al. [35] demonstrated that a lack of CD55 in a murine RA model was associated with a reduced severity of synovitis. These seemingly contradictory findings may be explained when accounting for varied CD55 activity depending on the surrounding cells [32].
The most recent studies suggest that CD55 may be a positive regulator of tumor formation and malaria, while being a negative regulator of autoimmune and demyelinating conditions, paroxysmal nocturnal hemoglobinuria, and protein losing enteropathy [32].
Study limitations
This study needs further processing of results in relationship with clinical data. The relatively small study group and control group limited the value of a statistic analysis, what positioned the work as an observational study. An additional limitation was that only one pathologist assessed the location and intensity of IHC reactions.
Conclusions
Our study demonstrated the absence of complement-cascade proteins (C1q, MAC) in the salivary glands of pSS patients, which indicated a lack of local complement activation via the classical pathway and the observed gland tissue damage being due to a mechanism other than MAC-induced cytolysis.
The observed differences in the expression of complement regulatory proteins in pSS, non-specific sialadenitis, and normal salivary glands in the absence of complement cascade activation may indicate that alternative functions of these regulatory proteins (e.g. modulation of cell-mediated response) may be of greater significance in pSS.
Low MCP expression in the salivary glands of patients with pSS, in comparison with that in the salivary glands in non-specific sialadenitis and that in normal salivary glands, may suggest altered modulation of cell-mediated immunity in the autoimmune process of pSS. Moreover, the differences in the location and intensity of protectin (CD59) expression in these three types of salivary gland samples indicates a possibility of reducing the proinflammatory effect of protectin in pSS.