Introduction
Menopause is a natural result of ageing in women and the end of the reproductive period which on average occurs at 51 years old [1, 2]. The menopausal period is associated with a decrease in ovarian follicles, cut-off menstruation and a dramatic decrease in serum levels of estradiol (E2) steroid hormone [1]. Researchers have shown that both premature and early menopause are associated with many negative neurological results. In the light of this information studies from a Mayo clinic cohort study of oophorectomy and ageing suggest a increased risk of cognitive impairment or dementia in women who underwent oophorectomy before menopause [3, 4]. These negative results were thought to be the effect of long-term and premature deficiency of E2. In a study that supported this finding, there was not increased risk of cognitive impairment and dementia in women who underwent bilateral oophorectomy before the age of 49 years but who had received estrogen treatment compared to women who underwent bilateral oophorectomy at the age of 50 years or more [2]. However, it is still not clear how E2 deficiency contributes to the deterioration of neurological functions. In experimental menopausal animal models, long-term E2 deficiency has been reported to have negative effects on neuroprotective effect of E2 on the brain in the hippocampal CA1 region. Nevertheless, while studies have focused on the CA1 region of the hippocampus in long-term E2 deficiency, the hippocampal CA3 region has been shown to be more affected by ischemic damage [5].
Recently, E2 has been accepted as a conditional neuroprotector [6]; that is to say, E2 protects neurons under a specific condition rather from any situation. An important effect of E2 is an antioxidant activity in which the phenolic portion of the structure of E2 clears the free radicals [7, 8].
The transient receptor potential M2 (TRPM2) channel is distributed in different parts of the mammalian brain mainly including the hippocampus, cortex, thalamus, midbrain and medulla oblongata; this channel is also found abundantly in neurons and microglia [9, 10]. High TRPM2 expression mediates entrance of extracellular calcium and induces cell death. TRPM2 plays important roles in many diseases such as traumatic brain injury [11], cerebral ischemia [12], cancer, inflammation and neurodegenerative diseases [9].
The dominant muscarinic acetylcholine receptors (CHRMs) in the central nervous system are the M1 subtype, which is expressed postsynaptically in the cortex, hippocampus, striatum, and thalamus [13]. Immuno-precipitation studies have shown that the M1 receptor population is dominant, although the hippocampus expresses all muscarinic receptor subtypes [14]. For this reason we have chosen to work with the muscarinic acetylcholine receptor 1 (CHRM1) receptor subtype. Pharmacological evidence [15] suggests that CHRM1 mediates the effects of acetylcholine, which plays an important role in cognitive processes, on cognition [16], hippocampal synaptic plasticity [17] and neuronal excitability [18]. The hippocampal estrogen modulates synaptic plasticity associated with memory at a slow but also rapid rate [19]. Furthermore, estrogen receptors are co-localized with CHRMs and nicotinic receptors in experimental rat hippocampal neurons [20].
The aim of the study was to investigate the association of TRPM2 and CHRM1 activity with the memorial functions that are deteriorated in the surgical menopause.
Material and methods
A total of 14 female Sprague-Dawley rats weighing between 200 and 220 g were housed in cages of 7, with 12-h cycles of light and darkness and were fed with standard pellet food and tap water.
Ovariectomy procedure
The abdomen was cleaned with 10% povidone-iodine solution and 14 rats were randomly and prospectively divided into 2 groups, each containing 7 rats. For the anesthesia, ketamine (Ketalar, Eczacıbaşı Warner-Lambert, Istanbul, Turkey) 30 mg/kg/im and xylazine hydrochloride (Rompun, Bayer, Istanbul, Turkey) 2 mg/kg/i.m. were administered.
G1 (n = 7): Sham group. The group of rats whose abdomen was opened and closed without any interventions.
G2 (n = 7): Surgical menopause group. The group whose abdomen was entered to perform bilateral ovariectomy.
The abdominal layers and the skin were closed with 3/0 silk. The rats were kept in cages seven by seven separately, until the end of the experiment. Zheng et al. [21] reported that ketamine induces hippocampal neurotoxicity by increasing apoptosis. For this reason to create the same cerebral effects by anesthesia in both groups, the anesthetic agent was given to the sham group and abdomen was opened and closed.
Experimental groups
For 14 days after the ovariectomy procedure, during the training and practice period, the rats were kept in the same room in polycarbonate cages (50 cm × 30 cm × 20 cm) having grates on the upper side. The circadian rhythm was set for 12 h of light and 12 h of darkness. The sawdust at the bottom of the cages was renewed twice each day. In order to recognize its smell, cheese pieces were added to the pellets they consumed.
Test procedure
Labyrinth arrangement as used by Pitten et al. [22] was used for the test apparatus. In our modified arrangement, an open-topped labyrinth in 100 × 100 × 20 cm dimensions containing chambers and corridors produced with wooden partitions was used. Above 3 of these chambers, there was an opening so that the rats may pass and enter into the partitions. The raw material of the labyrinth was formica-covered chipboard. The starting and finish points of the labyrinth were determined. A piece of cheese was inserted into the target point for the rats to find. The time taken to find the cheese was recorded and the obtained data were analyzed statistically.
Educational period
The rats in both groups were put on the starting point once a day for 5 days to become accommodated to the labyrinth. After this accommodation period, cheese was inserted at the target point for 8 days and the rats were helped to find it by an observer. In the next 8 days, the time taken to find the cheese was recorded. During this period, all rats were put in the labyrinth after they were numbered one by one and time was also given to the rats to eat the cheese. The required time for each rat was recorded with a chronometer. Before putting the next rat to the test, urine or stool that could produce an odor were cleaned from the labyrinth using a wet sponge. After the place was completely dried, the period of the next rat was started.
Second surgical procedure
The day after the educational period was completed, the rats were decapitated under high dose anesthesia. After the brain tissues of the rats were quickly removed, the brain was divided into right and left hemispheres from the middle line. The right hemisphere was placed in 10% formol for histopathological and immunohistochemical examination. The hippocampus of the left hemisphere was removed and stored at –80°C in an eppendorf tube.
RNA isolation, complementary DNA (cDNA) synthesis and quantitative real-time PCR (qRT-PCR) analysis
Total RNA isolation from hippocampal tissue was performed with Tri Reagent according to the protocol supplied by the manufacturer (Bioshop, Canada). The RNA purity and concentration were measured using a MaestroNano Spectrophotometer at 260 nm (MaestroGen, Taiwan). cDNA amplification was carried out in a thermal cycler (Applied Biosystems Veriti Thermal Cycler, CA, USA) using the Applied Biosystems High Capacity RNA-cDNA Kit (Applied Biosystems, CA, USA). Gene expression levels were measured with the Applied Biosystems 7500 Real-Time PCR system. Amplification was performed by using gene specific primers TRPM2 (Assay ID. Rn01429410_m1, Qiagen) and CHRM1 (Assay ID. Rn00589936_s1, Qiagen)/probes and TaqMan Master Mix (Applied Biosystems, Universal TaqMan Gene Expression Master Mix, CA, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Assay ID. Rn01775763_g1, Qiagen) was used as an endogenous control gene (housekeeping). At the end of qRT-PCR analysis, the 2–ΔΔCT method was used to calculate the differences in gene expression [23].
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining Sections of 5–6 mm thickness obtained from paraffin blocks were taken to glass slides with polylysine. In accordance with the manufacturer’s instructions, cells undergoing apoptosis were established using the ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (Chemicon, cat no: S7101, USA). Tissues deparaffinized with xylene were treated with graded alcohol and washed with phosphate buffered saline (PBS). Incubated with 0.05% proteinase K, tissues were then incubated for 5 min with 3% hydrogen peroxide in order to prevent endogenous peroxidase activity. After washing with PBS, tissues were incubated for 6 min with equilibration buffer and again incubated at 37°C in a humid environment with study solution (70% ml Reaction Buffer + 30% TdT Enzyme) for 60 min. Maintained in Stop/Wash Buffer for 10 min, tissues were then treated with anti-digoxigenin peroxidase for 30 min. Apoptotic cells were imaged with diaminobenzidine (DAB) substrate. Sections that were contrast stained with Harris hematoxylin were shut down with appropriate solution. The preparations were examined and evaluated under a test microscope (Olympus BX-50) and then photographed. In the evaluation of TUNEL staining, kernels stained in blue with Harris hematoxylin were assessed as normal, and cells demonstrating brown nuclear staining were established as apoptotic. Extensity of the staining was taken as the basis when evaluating TUNEL staining. In each case by microscopic evaluation the apoptotic index was determined. In sections, TUNEL-positive cells and total cells were counted in 10 random areas, under 40× magnification objective. The apoptotic index was calculated by the formula: 100× (mean number of TUNEL positive cells in 10 random fields)/(mean number of total cells in 10 random fields) and statistical analyses were performed [24]. The preparations were examined under a test microscope (Olympus BX-50) and then photographed.
Sampling and determination of malondialdehyde levels
The obtained hippocampal tissues were cleaned with cold (+4ºC) 0.9% sodium chloride and dried with absorbent paper. Tissues were then homogenized for 3 min at 16000 revolutions/min in 0.01 M phosphate-buffered saline solution (1 : 9 weight/volume) using a homogenizer (Ultra Turrax Type T25-B, IKA Labortechnic, Staufen im Breisgau, Germany). Homogenization was performed on ice, with supernatants obtained by centrifugation at 5000 g for 1 h (at +4ºC). Levels of MDA were assayed spectrophotometrically by the Ohkawa method [25] in terms of nmol per g wet tissue. Malondialdehyde levels were determined based on the coupling of malondialdehyde with thiobarbituric acid at +95°C. Determination of lipid peroxidation depends on the spectrophotometric measurement at 532 nm of the pink complex obtained by incubation of 0.8% thiobarbituric acid with tissue homogenate in a boiling water bath for 1 h under aerobic conditions at pH 3.5. For the measurements, we used standard 1,1,3,3-tetraethoxypropane.
Immunohistochemistry (IHC)
Deparaffinized tissues were passed through a graded alcohol series and boiled in citrate buffer solution at pH 6 in a microwave oven (750 W) for 12 min in order for antigen retrieval as defined by Atilgan et al. [26]. In order to prevent surface staining, after treatment with Ultra V Block (TA-125-UB, the Lab Vision Corporation, USA) solutions, the tissues were incubated with primary antibodies for 60 min (CHRM1 was purchased from Boster (Cholinergic receptor, muscarinic 1, catalog number: PA2202, Boster, 3942 B Valley Ave, Pleasanton, CA, 94566) and TRPM2 was purchased from Abcam (Rabbit Anti-TRPM2 antibody, ab101738, Abcam, Cambridge, UK)). After the application of primary antibodies, tissues were incubated with secondary antibodies (30 min) (biotinized anti-mouse/rabbit IgG, Diagnostic BioSystems, KP 50A, Pleasanton, USA), streptavidin alkaline phosphatase (30 min) (TS-060-AP, the Lab Vision Corporation, USA) and Fast Red Substrate System (TA-125-AF, the Lab Vision Corporation, USA). Tissues that were exposed to a contrasting staining with Mayer’s hematoxylin were treated with PBS (phosphate buffered saline) and distilled water, and then closed with the appropriate shutdown solution. The prepared tissues were examined and evaluated under an Olympus BX50 microscope and photographed. Extent of staining was taken as the basis when evaluating the immunohistochemical staining. A histoscore was derived from the distribution (0.1 = ≤ 25%; 0.4 = 26–50%; 0.6 = 51–75%; 0.9 = 76–100%) and the intensity (0 = no staining; +0.5 = very little staining; +1 = little staining; +2 = medium staining; +3 = very strong staining) of the staining immunoreactivity (histoscore = distribution × intensity) [26].
Statistical analysis
The results were expressed as arithmetic mean ± standard deviation (mean ± SD). All analyses were performed with the program SPSS 22.0 for IBM Corporation Armonk, NY, USA. Kruskal-Wallis variance analysis was conducted, and then the Mann–Whitney U test was used for inter-group dual comparisons for parameters found as p < 0.05.
Results
Results of education and memory
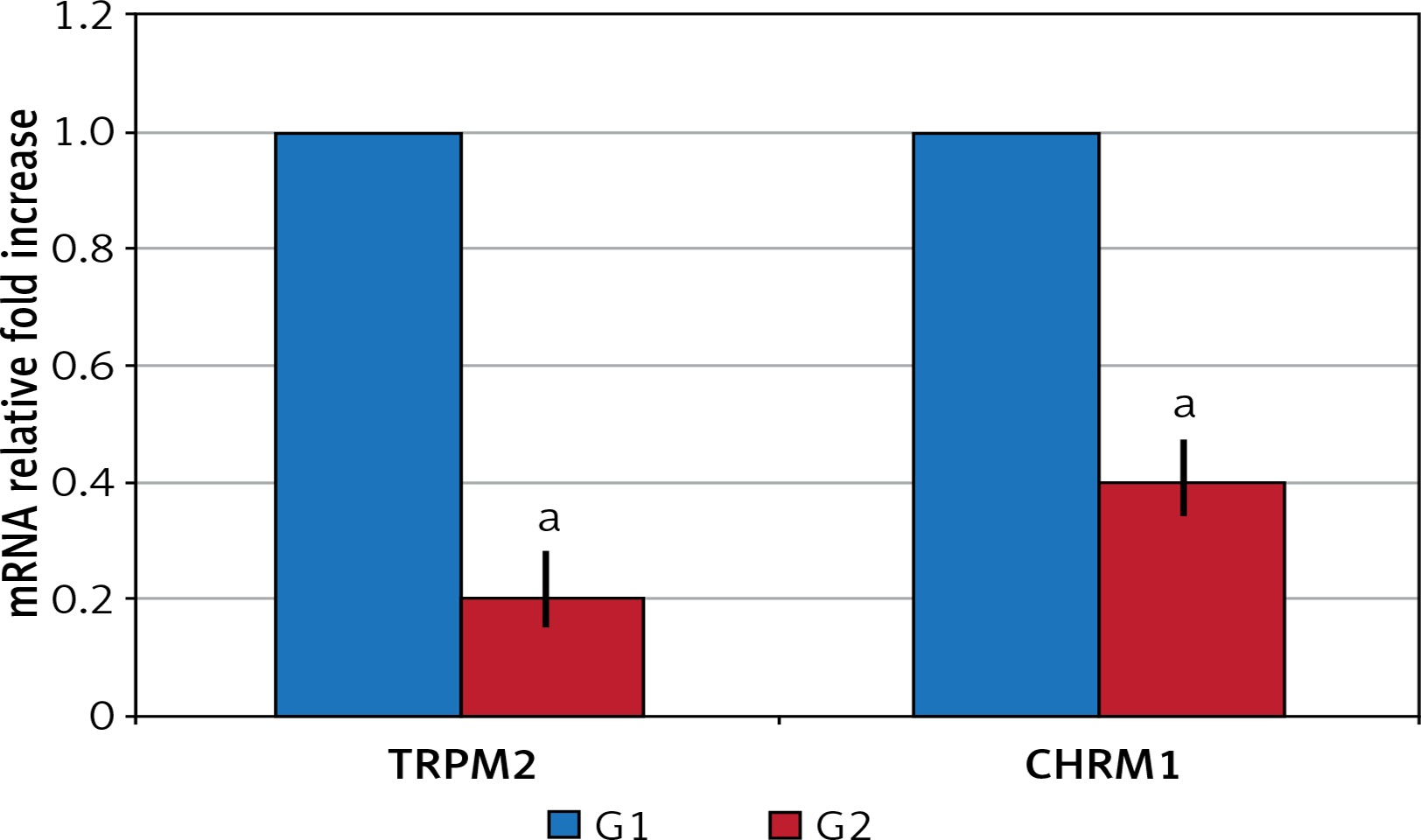
In the surgical menopause group, the mean time required for the rats to find the cheese was statistically significantly elongated (p < 0.05) (Table I). TRPM2 and CHRM1 gene expression: Significant down-regulation was observed in G2 compared to G1 in TRPM2 and CHRM1 gene expressions (p < 0.05) (Figure 1).
Table I
Time required for rats to find the cheese
| Groups | Time required to find the cheese [s] Mean ± standard deviation |
|---|---|
| G1 | 29.86 ±2.6 |
| G2 | 39.29 ±4.0a |
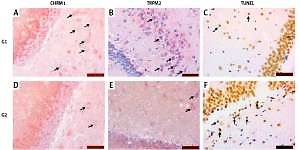
TRPM2 and CHRM1 immunoreactivity
TRPM2 and CHRM1 immunoreactivity were observed in hippocampus of the rats. When the hippocampal TRPM2 and CHRM1 immunoreactivity results were evaluated; there were a statistically significant decrease in G2 group compared with the G1 (p < 0.05), (Table II, Figure 2).
Table II
CHRM1, TRPM2 immunoreactivity and apoptotic histoscore (diffuseness × severity) and MDA levels
| Groups | CHRM1 | TRPM2 | TUNEL | MDA [nmol/ml] |
|---|---|---|---|---|
| G1 | 1.30 ±0.58 | 1.65 ±0.79 | 2.66 ±1.63 | 19.82 ±1.30 |
| G2 | 0.36 ±0.05a | 0.55 ±0.08a | 12.40 ±2.30a | 45.56 ±8.35a |
Figure 2
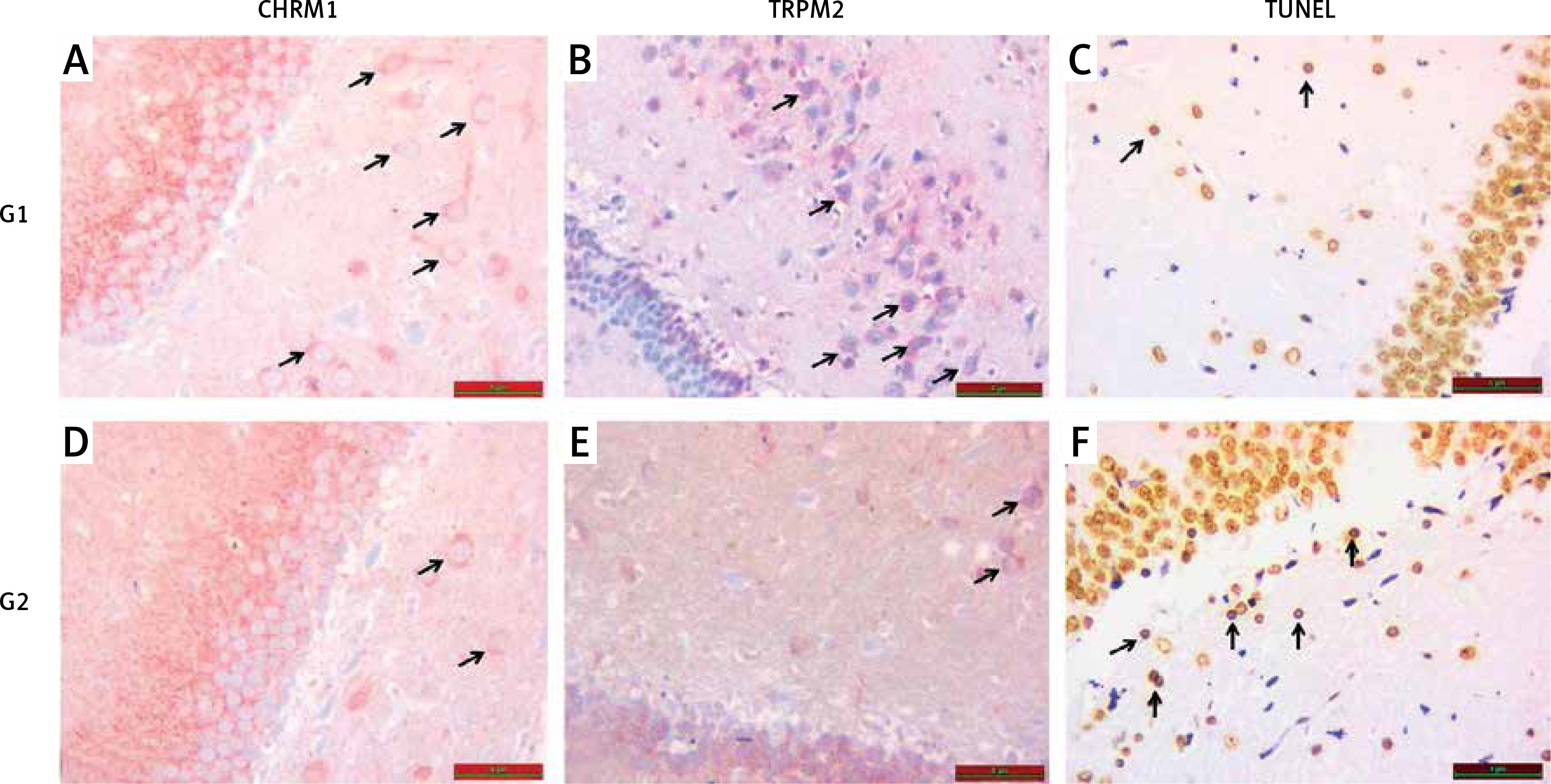
A – Increased chrm1 immunoreactivity staining in hippocampal area is shown in G1 relative to G2 (black arrow). B – Increased trpm2 immunoreactivity staining in hippocampal area is shown in G1 relative to G2 (black arrow). C – TUNEL staining in hippocampal area is lower in G1 compared with G2 (black arrow). D – CHRM1 immunoreactivity staining in hippocampal area is lower in G2 compared with G1 (black arrow). E – TRPM2 immunoreactivity staining in hippocampal area is lower in G2 compared with G1 (black arrow). F – TUNEL staining in hippocampal area is higher in G2 compared with G1 (black arrow)
TUNEL result
When compared with G1, an increase in hippocampal TUNEL positivity was significantly observed in G2 (p < 0.05), (Table II, Figure 2).
Spectrophotometric MDA results
When compared with G1, there was a significant increase in hippocampal MDA in G2 (p < 0.05), (Table II).
Discussion
In our study, we observed that surgical menopause increased apoptosis caused by ischemia-reperfusion injury in hippocampus, and decreased TRPM2 and CHRM1 activities and as a result caused impairment in learning and memory.
Oxidative stress induced by cerebral ischemia-reperfusion injury is considered to be the main event leading to neuronal death [27]. As a result of cerebral ischemia, ADPR production increases in the rat brain [28]. Activation of the enzyme ADPR pyrophosphatase by some stimulants such as ADPR and oxidative stress triggers the TRPM2 channel [29]. Therefore, in some studies, TRPM2 has been shown to mediate ROS-induced neuronal death. TRPM2 acts as a redox-sensitive Ca2+ transparent channel. Therefore, TRPM2 has been shown to play an important role in H2O2-induced neuronal death [30] in primary cultured neurons [31] and organotypic hippocampal culture [32]. The significant increase in hippocampal MDA levels in the surgical menopause group in our study is an indicator of ischemia reperfusion injury, because MDA is used as one of the markers of ROS levels in the body [33].
We also expected an increase in TRPM2 levels due to increased oxidative damage in G2, whereas by contrast we found down-regulation of TRPM2 gene expression and a significant decrease in immunoreactivity score. Independent of sex steroids, in women TRPM2-mediated injury mechanisms was found to be absent. Neither estrogen withdrawal nor androgen supplementation has been shown to induce TRPM2-mediated injury after experimental stroke in female animals [34]. Following cerebral ischemia, TRPM2-mediated neuronal damage is a specific cell death path for males [30, 35]. Ischemia was reported to have failed to provide adequate stimulation of the active TRPM2 channels in the female brain. The simplest explanation is the relative lack of PARP-mediated ADPR in the female brain following ischemia [34]. This information may also indicate why hippocampal TRPM2 activity in our menopausal group is not elevated. Similarly, in another study, it was observed that the level of TRPM2 and TRPV1 expression in the brain decreased with experimental brain injury [36].
Meng et al. [37] observed that the decrease in learning and memory functions in surgical menopausal rats was related to degenerative changes in the hippocampal neurons. Estrogen deficiency increased apoptotic cells and decreased anti-apoptotic protein Bcl2. Although the death of neurons by apoptosis is still not clear, it is suggested to be associated with activating intercellular pathways which will result in phagocytic cells to take in neurons [38, 39]. Chronic activation of the microglia is thought to induce inflammation that results in neuronal cell death [40]. The hippocampus has the ability to regenerate and it is susceptible to environmental stimuli including oxidative stress and Ca2+ entry [41]. Calcium flow due to TRPM2 activation has been shown to facilitate cell death pathways independently of apoptosis and caspase [42]. In a previous study it was found that TRPM2 increases as a result of glial activation at 1 and 4 weeks of ischemic damage and that TRPM2 was produced at high rates within a short period of time, suggesting a possible role in ischemia pathophysiology [43]. During the apoptotic process, the cell pH decreases rapidly, calcium pump failure results in continuous calcium entry into the cell, and calcium increases in the cell [44]. Extracellular acidic pH has been shown to strongly inhibit the TRPM2 channel [45]. This may be another reason for the decrease in TRPM2 activity in our menopausal group. Interestingly, ischemia-activated cell death pathways are also different between males and females [46]. Caspase-dependent cell death pathways are predominant in women [47], whereas oxidative stress-dependent activation and apoptosis inducing factor stimulation of poly (ADP-ribose) polymerase (PARP) are involved in men [48, 49]. Bal et al. [50] reported that inactivating TRPM2 ion channels would modulate the neuronal activity in the central audio path by reducing neural excitability, presumed to cause an increase in inhibitory signals in auditory nuclei leading to decreased tinnitus-producing signals. This suggests that a brain-protective mechanism against apoptosis in our menopausal group may work to prevent the apoptotic cascade by inhibiting TRPM2. Perhaps the peripheral estrogen conversion [51] in the surgical menopause group protects the brain from partial ischemia with an antioxidant effect, which suppresses TRPM2 levels.
Improvement of the cholinergic system leads to improvement of cognitive functions. It is known that especially a high ACh level improves learning and memory [52]. Cholinergic receptors are vital members of the cholinergic system and the role of CHRMs (M1-M5) in cognition has already been established [53]. M1 receptor stimulation leads to the development of cognition [54]. Estrogen receptors are co-localized with CHRMs and nicotinic receptors in rat hippocampal neurons [20]. It has previously been shown that E2 modulates the expression and function of CHRMs in the rat hippocampus [55]. Several mechanisms may contribute to the effects of estrogen on the hippocampus. Firstly, E2 can directly enhance the muscarinic acetylcholine receptor gene expression in the hippocampus. Alternatively, E2 can indirectly stimulate gene expression of CHRMs by increasing the expression of growth factors in the hippocampus [14].
We found that there was down-regulation in CHRM1 gene expressions in the group to which we applied surgical menopause. Immunohistochemically, there was also a significant decrease in CHRM1 staining scored in the menopause group. The rapid decline in sex steroids [56] and elevated oxidative damage may explain down-regulation and decreased immunoreactivity in CHRM1. Ischemia reperfusion injury stimulates some harmful neurobiological mechanisms such as inflammation and oxidative damage. This can lead to serious impairment of cholinergic functions, neurological damage, and consequently impaired memory performance [57, 58]. Ischemia reperfusion injury in our menopausal group may explain the decrease in CHRM1 activity and as a result the cause of learning and memory impairment.
The cognitive effects of androgens probably depend on a woman’s background hormone profile in the postmenopausal period and this should also be taken into account when interpreting the effects of ovarian hormones on cognitive outcomes [16]. Androstenedione can be aromatized to estrone, an estrogen that was demonstrated to disrupt memory in an ovariectomized mouse model [51]. Taken together, these new findings indicate that serum estrogen levels alone do not necessarily dictate or predict cognitive outcomes. Memory performance is part of a complex and interrelated system involving many cellular and molecular mechanisms [16]. Activation of TRPM2 causes Ca2+ entry along the plasma membrane and Ca2+ release from lysosomes [59], whereas activation of CHRMs changes the activity of membrane ion channels [60]. We evaluated TRPM2 and CHRM1 activities together because of these similar activities and since both were associated with ischemia reperfusion injury. Increases in intracellular Ca2+ levels have been shown to disrupt activation of ion channels and gap junction communication [59]. This information may be a guide for understanding the reduction in TRPM2 and CHRM1 activity with a decrease in learning and memory in surgical menopause. However, further experimental physiological studies are needed to explain this relationship.
The limitations of our study were as follows: this was an experimental study with a limited number of cases, and hippocampal pH values were not studied. In our study, TRPM2 and CHRM1 activities were studied in surgical menopause for the first time in the literature.
In conclusion, surgical menopause leads to an increase in apoptosis in the hippocampus, and decrease in TRPM2 and CHRM1 activities may cause deterioration in learning and memory.