Introduction
β-Thalassemia major (β-TM) is a common congenital blood disorder caused by mutations in the β-globin gene [1]. Reduced or absent β-globin expression leads to an imbalance of the α-globin, and free α-globin molecules form insoluble aggregates that damage and destroy erythroid precursors in a process termed ineffective erythropoiesis [2]. During the last years some studies indicating the role of the complement system in the etiopathogenesis of hemolysis observed in β-TM were published [3]. Hemolysis induced by activation of the complement system is prevented by complement regulatory proteins. Decay accelerating factor (DAF, CD55), membrane inhibitor of reactive lysis (MIRL, CD59), and complement reception 1 (CD35) are among these proteins. CD55 and CD59, which are glycosylphosphatidylinositol anchored, inhibit formation of the C3 convertases and prevent the terminal polymerization of the membrane attack complex on the cell surface of erythrocytes. CD35 is also involved in the regulation of C3 fragment deposition. The absence of these proteins thus accounts for the increased susceptibility of erythrocytes to complement lysis [4–6].
The majority of β-thalassemia patients depend on regular blood transfusions combined with iron chelation therapy for their survival. If the bone marrow becomes insufficient to counteract the anemia, the spleen works as an extramedullary organ and hypersplenism develops in β-TM. Hypersplenism caused by extramedullary hematopoiesis necessitates splenectomy [7]. The spleen is crucial in regulating immune homoeostasis through its ability to link innate and adaptive immunity leading to protection against infections [8]. The most serious problem is the infection (Streptococcus pneumonia, Haemophilus influenza, Neisseria meningitides) in splenectomized patients. The role of the complement system in susceptibility to infections has been shown in previous studies [9]. In this study we investigated how splenectomy affects complement regulatory proteins on erythrocytes in β-TM patients. Then we analyzed CD35, CD55, and CD59 levels on erythrocytes in β-TM by flow cytometry.
Material and methods
Subjects
This study was carried out on 45 β-TM (22 β-TM with splenectomy and 23 β-TM without splenectomy) patients and 17 healthy volunteers as control subjects. β-TM patients were followed up at the Thalassemia Center of the Antalya Education and Research Hospital, Antalya, Turkey. These patients were seen during their routine follow-up visits at the outpatients clinic and a detailed history was taken from all patients regarding age, time of the first blood transfusion, time of the last transfusion, frequency of transfusions and other information that concern the patient’s status. β-TM patients received blood transfusion according to their blood counts (patients without splenectomy: 1–2 weeks, patients with splenectomy: 3–4 weeks intervals). Peripheral blood samples were collected in K2-EDTA vacutainer tubes before receiving the transfusion. The analyses were carried out promptly including a complete blood count (CBC) that was performed for all patients and control subjects using a hematology analyzer (Coulter LH 780 Analyzer, Beckman Coulter). This study was performed with the approval (Number: 096/2015) of the ethics committee of the Education and Research Hospital Antalya, Turkey. All subjects were informed about the objectives and procedures of the study, and informed consent was obtained from each subject.
Flow cytometric analysis of CD55, CD59 and CD35 on the cell membrane of erythrocytes
The EDTA blood sample was processed within an hour. The blood sample was diluted with phosphate-buffered saline (PBS) to achieve 10 000 RBC/µl. Then 100 µl of diluted blood was transferred to a polystyrene tube and to each polystyrene tube, 10 µl of monoclonal antibodies against CD55 (PE Mouse Anti-Human), CD59 (FITC Mouse Anti-Human), and CD35 (PE Mouse Anti-Human) were added. After 30 min incubation in the dark at room temperature, samples were washed twice by PBS. Finally, the pellet was resuspended in 0.5 ml of PBS and cells were analyzed in the flow cytometer (FACS Canto II, Becton-Dickinson).
Statistical analysis
For data description, maximum, minimum, mean, standard deviation (SD) and median were tabulated. The Shapiro-Wilk test was used to verify the normality of the distribution of continuous variables. Statistical analysis of clinical data between two groups (control vs. patients) consisted of the unpaired t-test for parametric data and Mann-Whitney U test analysis for nonparametric data. Analyses were performed with PASW 20 (SPSS/IBM, Chicago, IL, USA) software and a two-tailed p-value less than 0.05 was considered statistically significant.
Results
The study included a total of 45 patients with β-TM, divided into two groups according to the absence (group 1: 22 β-TM with splenectomy) or presence (group 2: 23 β-TM without splenectomy) of the spleen. Seventeen healthy volunteers (group 3: controls) were included in the present study. There was no statistically significant difference regarding age and gender between patient and control groups. The hematological parameters of the patients and controls are summarized in Table I.
Table I
Hematological parameters of group 1 and 2 compared to group 3
| Parameter | Group 1 Mean ± SD | Group 2 Mean ± SD | Group 3Mean ± SD |
|---|---|---|---|
| RBC count [× 1012/l] | 3.27 ±0.28 | 3.18 ±0.45 | 4.83 ±0.4* |
| Hb [g/dl] | 8.92 ±0.79 | 8.57 ±0.81 | 14.3 ±1.35* |
| MCV [fl] | 82.92 ±2.65 | 80.46 ±4.76 | 87.36 ±2.62* |
| MCH [pg] | 27.38 ±1.68 | 27.29 ±1.88 | 29.65 ±1.23* |
| MCHC [g/dl] | 33.02 ±1.52 | 33.92 ±0.99 | 33.97 ±0.86 |
| RDW (%) | 18.28 ±5.17 | 18.28 ±6.72 | 13.32 ±0.69* |
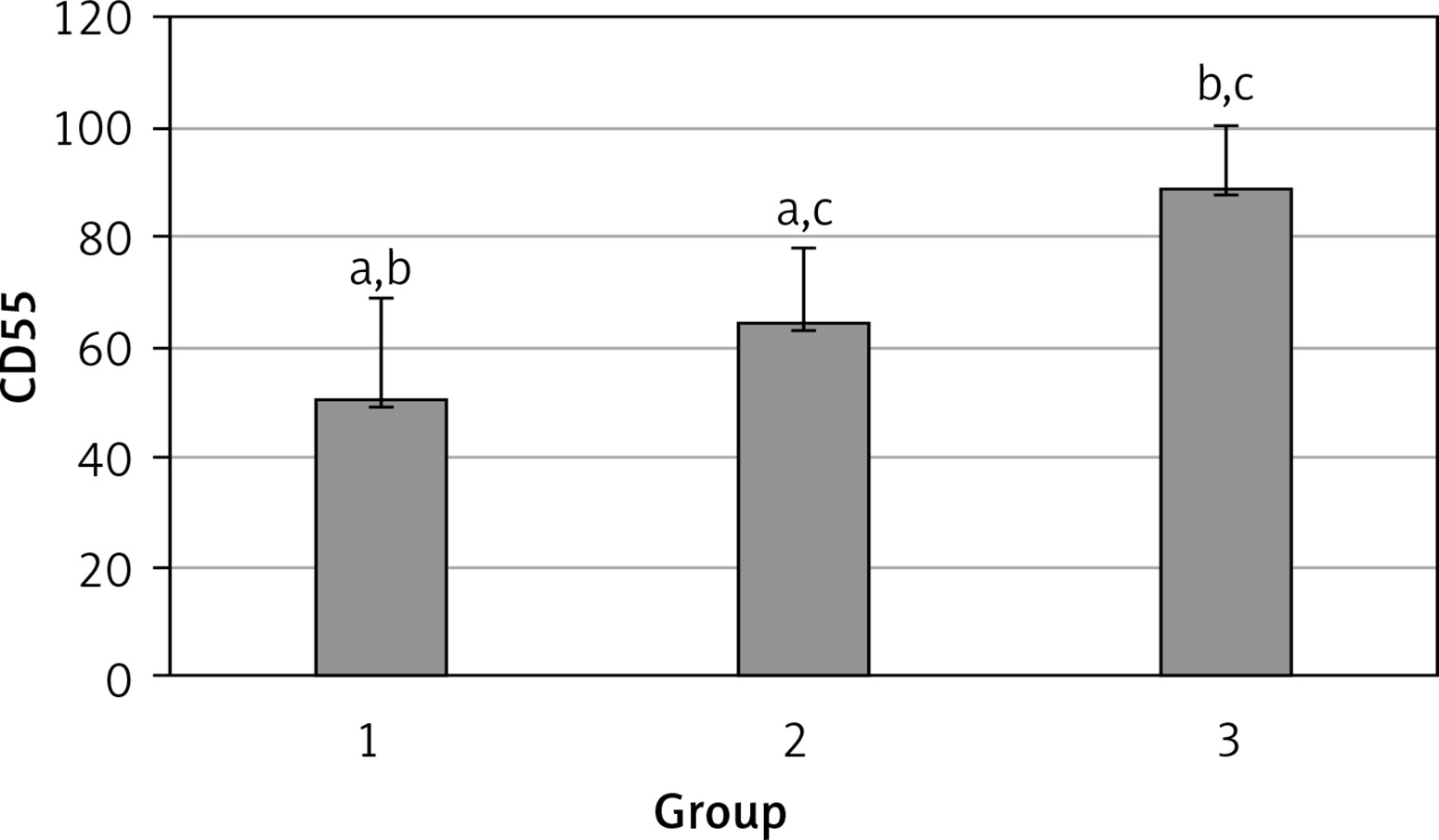
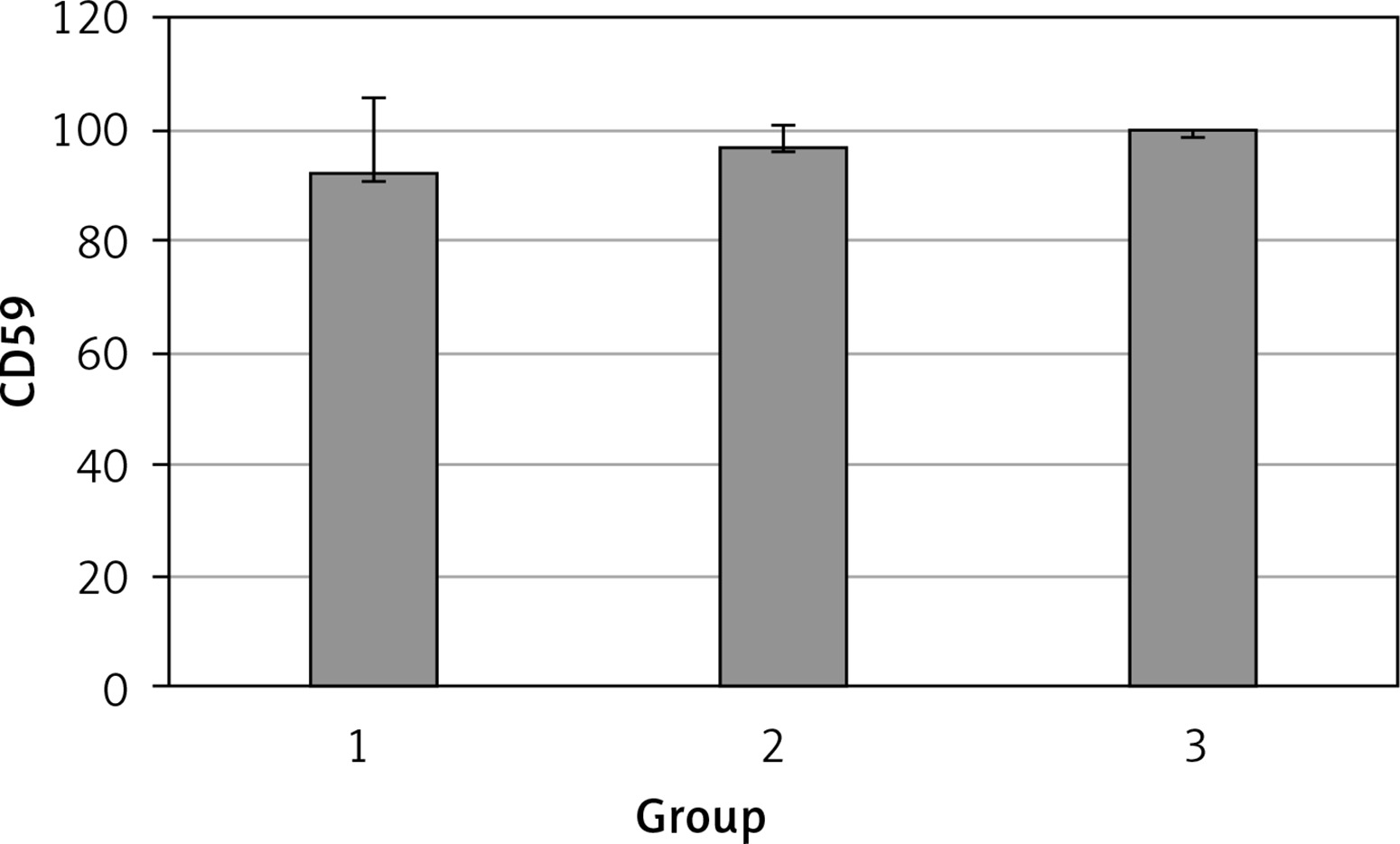
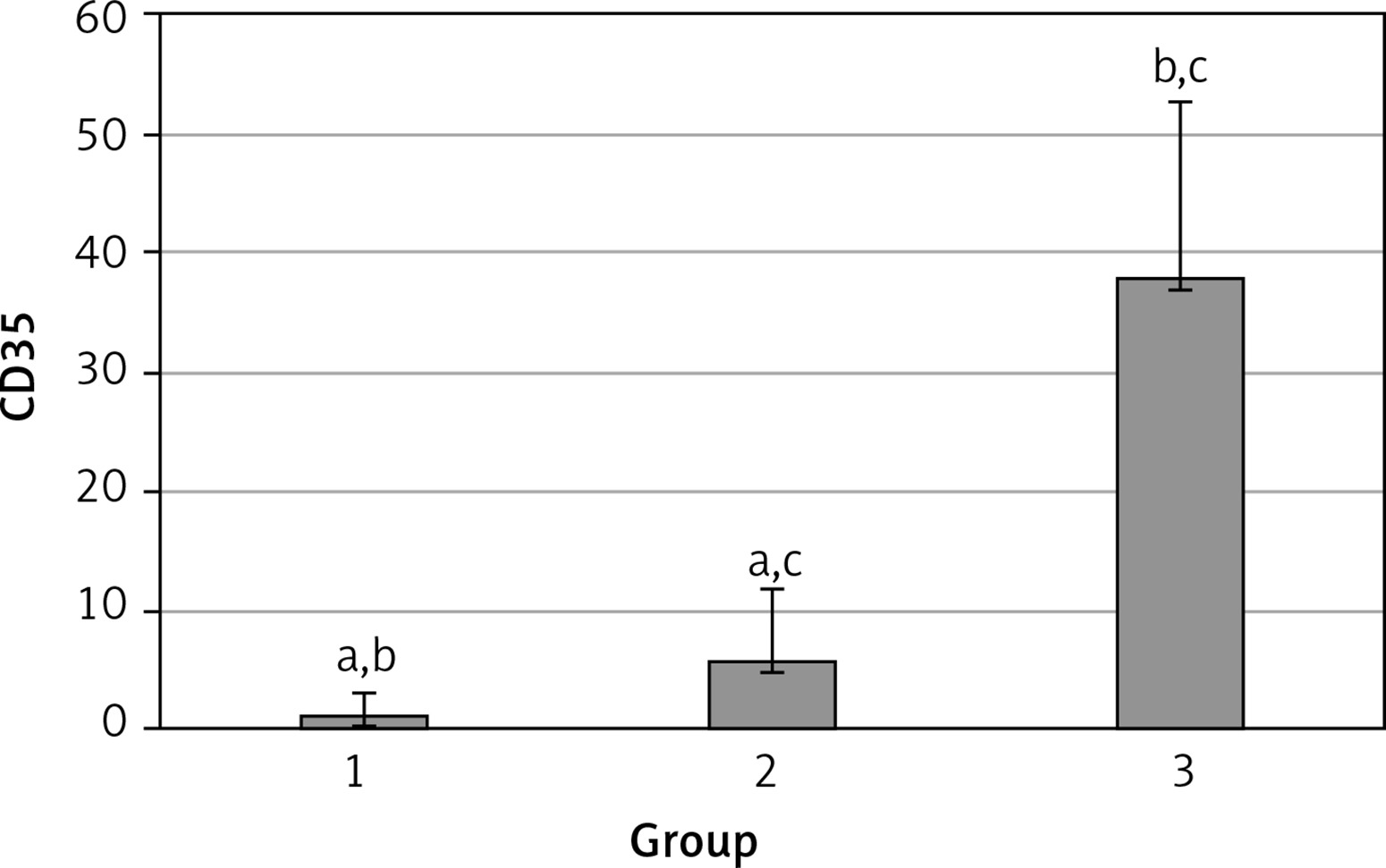
The levels of CD55, CD59, and CD35 expression on peripheral blood erythrocytes by flow cytometric analysis were compared between groups 1 and 2, as shown in Table II. The overall mean percentage of CD55-positive erythrocytes of group 1 (50.72 ±18.67%) was significantly lower than in group 2 (64.36 ±13.98%) and group 3 (89.03 ±11.75%) (p < 0.05). Also the CD55-positive erythrocyte percentage of group 2 was significantly lower than in group 3 (p < 0.05) (Figure 1). The overall mean percentage of CD59-positive erythrocytes of patients was not significantly different in all groups (group 1: 92.3 ±13.65, group 2: 96.85 ±4.18, group: 3 99.87 ±0.18) (Figure 2). The overall mean percentage of CD35-positive erythrocytes of group 1 (1.32 ±1.74%) was significantly lower than in group 2 (5.75 ±6.09%) and group 3 (38.03 ±14.72%) (p < 0.05). Also the CD35-positive erythrocyte percentage of group 2 was significantly lower than in group 3 (p < 0.05) (Figure 3).
Table II
Levels of CD55, CD59, and CD35 expression for group 1 compared to group 2
| CD | Group 1 | Group 2 | P-value | ||
|---|---|---|---|---|---|
| Mean ± SD | Median (range) | Mean ± SD | Median (range) | ||
| CD55 | 50.72 ±18.67 | 54.85 (7.4–76.8) | 64.36 ±13.98 | 99.25 (51.4–97.4) | < 0.05* |
| CD59 | 92.3 ±13.65 | 97.4 (54–99.4) | 96.85 ±4.18 | 96.8 (89.6–100) | > 0.05 |
| CD35 | 1.32 ±1.74 | 0.7 (0–7.3) | 5.75 ±6.09 | 2.3 (0–24.6) | < 0.05* |
Discussion
β-TM is due to impaired production of β-globin chains, leading to a relative excess of α-globin chains. Excess α-globin chains are unstable, incapable of forming soluble tetramers on their own, and precipitate within the cell, leading to a variety of clinical manifestations [10]. The direct effects of β-TM on other organs and tissues are due to the deleterious effects of the profound anemia, the byproducts of hemolysis, and the intramedullary and extramedullary expansion of erythroid marrow progenitors [11]. Massive splenomegaly develops early in the course of β-TM due to increased erythrocyte destruction and the presence of splenic extramedullary hematopoiesis [12].
During the last years the relationship between complement system and hemolysis has been under investigation. It has been emphasized that especially regulatory proteins in the complement system have an important role [13]. Complement regulatory proteins prevent complement damage to normal host tissue and fluid-phase activation. Deficiencies of control proteins lead to excessive complement activation and significant morbidity and mortality [14]. In this study we investigated how splenectomy affects the level of expression of complement regulatory proteins, CD35, CD55, and CD59 on erythrocytes in β-TM patients.
Most peripheral blood cells express CD35 (complement receptor 1 (CR1)). The function of CD35 depends in part upon the type of cell on which it is expressed. Erythrocytes express approximately 500 copies of CD35 per cell. The primary function of CD35 on erythrocytes is to clear circulating immune complexes. CD35 plays a critical role in the clearance of C4b- and C3b-coated particles (e.g., immune complexes) on which the complement system has been activated. CD35 binds to intravascular immune complexes and serves as a vehicle (taxi) that transports immune complexes to the liver and spleen. Hepatic and splenic macrophages then “clear” such immune complexes by stripping them from the erythrocytes, destroying the antigen (often viruses or bacteria), and facilitating an immune response (antigen presentation). The erythrocyte may then return to the circulation for another round of immune complex clearance [15, 16]. In our study we found that the level of CD35 expression on the erythrocytes was significantly lower in β-TM patients compared to healthy controls. Similarly, the level of CD35 expression on the erythrocytes of splenectomized patients was significantly lower than in non-splenectomized patients.
Erythrocytes are protected from complement lysis by GPI-anchored proteins (CD55, CD59) on their surface that block complement activation. Intravascular hemolysis is normally blocked by CD59 (membrane inhibitor of reactive lysis (MIRL)), which prevents the final stage of complement assembly, production of the membrane attack complex (MAC) that forms a pore in the target cell. Extravascular hemolysis is normally blocked by CD55 (decay accelerating factor – DAF), which prevents assembly of the C3 and C5 convertases upstream of MAC formation in the complement cascade. CD55 protects erythrocytes from hemolysis by inhibiting C3 convertases such as the C4b2a complex, which enzymatically cleaves and activates C3. Accumulation of C3 on the erythrocyte surface in the absence of CD55 identifies (opsonizes) erythrocytes for reticuloendothelial destruction. Intravascular hemolysis occurs in PNH because erythrocytes lack the GPI anchor required to attach CD59 to their surface. Extravascular hemolysis also occurs in PNH, due to complement-mediated destruction of erythrocytes by reticuloendothelial macrophages in the liver and spleen [17–19]. In our study we found that the level of CD55 expression on the erythrocytes was significantly lower in β-TM patients compared to healthy controls. But there was no difference regarding CD59 levels between the two groups. CD55 levels of patients with splenectomy were significantly lower compared to non-splenectomized patients. Detection of no difference between β-TM patients and healthy controls regarding CD59 levels indicates that there are not intravascular hemolytic processes in β-TM patients. Presence of a low level of CD55 in β-TM patients indicates the presence of extravascular hemolysis.
In recent years studies about the complications of splenectomy affecting erythrocytes draws special attention among studies related to hemolytic anemias. Ying et al. investigated the effects of splenectomy on the erythrocyte membrane of hereditary spherocytosis patients by atomic force microscopy. Their observations demonstrated that splenectomy does not improve the underlying genetic defects in the RBC membrane. In fact, after splenectomy the underlying structural abnormalities in the RBCs of HS patients are even more obvious, as suggested by significantly decreased cells membrane roughness and smaller cell surface particles size [20]. Ibrahim et al. investigated the phosphatidylserine (PS) exposure on the erythrocyte membrane in β-thalassemia patients. They found that erythrocyte PS exposure was significantly higher in splenectomized β-TM patients compared with nonsplenectomized β-TM patients [21]. Lal et al. reported that splenectomy and iron overload have an additive effect on increasing the frequency of mitochondrial DNA deletions (ΔmtDNA497) in β-Thal patients [22]. Splenectomy results in damage to both the erythrocyte membrane and mitochondrial DNA as shown in these studies. In our study we demonstrated that both CD35 and CD55 are lower in splenectomized patients compared to non-splenectomized patients. Malik et al. emphasized a relationship between CD35 protein and mitochondrial DNA in a study in which the effects of ultraviolet radiation on age-related patients with macular degeneration were investigated. Also it was found that there was no change in the level of CD59 in this study [23]. Therefore we thought that the reason for low levels of CD35 and CD55 expression on erythrocyte membrane in splenectomized β-TM patients may be due to increased mtDNA and membrane damage.
We think that another reason for the low levels of both CD35 and CD55 in splenectomized patients may be transfusion frequency. Whereas splenectomized patients need 1–2 units of erythrocytes per 3–4 weeks, non-splenectomized patients need 1–2 units of erythrocytes per 1–2 weeks. Actually according to our hypothesis CD35 and CD55 levels of β-TM patients should be much lower than these values, and regulatory proteins detected in samples obtained from patients should belong to erythrocytes of healthy blood donors. This hypothesis explains the presence of higher levels of CD35 and CD55 in patients with a spleen, who need frequent transfusions, compared to splenectomized patients, because these patients have higher amounts of healthy erythrocytes due to transfusions.
The results will facilitate a better understanding of the impact of splenectomy on RBCs of β-TM patients. Lower levels of CD35 and CD55 on erythrocytes increase erythrocyte destruction. Increased erythrocyte destruction and iron deposition in organs due to deficiency of these regulatory proteins may be the underlying mechanism of organ damage (cardiac failure, hepatomegaly, and renal problems) developing in β-TM patients.
In the future measurement of CD35 and CD55 levels on erythrocytes may be a parameter used for treatment planning. There is a need for further studies investigating possible reasons for low levels of CD35 and CD55 in β-TM patients.