Introduction
Alzheimer’s disease (AD) is known to lead to some well-characterized pathological changes, such as the extracellular accumulation of amyloid plaques, intra-neuronal presence of neurofibrillary tangles, glial hypertrophy, and neuronal death [1–3]. The disease is mainly associated with neuronal damage; however, there is evidence that myelin fibers are also damaged in AD. The myelin pathology in patients with AD has not been widely studied so far, even though Alois Alzheimer described myelin disruption in AD as early as in 1911 [4, 5]. It is unclear why the phenomenon of myelin impairment has been forgotten for over 100 years. Classical neuropathological changes in AD, e.g. amyloid plaque deposition and the presence of neurofibrillary tangles in the brain, are responsible for neuronal damage and synapse loss, but oligodendroglial degeneration and myelin impairment have also been observed in the brains of AD patients [6–9].
The myelin sheath is a lipid-rich, multilamellar membrane wrapped around axons. Myelin impairment has been suggested to lead to neuronal dysfunction and to cognitive decline. Myelin is necessary for impulse propagation along axons. Additionally, there is evidence from neuroimaging and human brain postmortem studies that myelin damage may be associated with the presence of amyloid β (Aβ) plaques and tau hyperphosphorylation, which are both found in AD [6, 7, 10, 11]. What is interesting, the spread of AD pathology reflects the pattern of myelination in reverse [12]. Later myelinated brain regions, such as the temporal and frontal lobes, develop AD pathology before early myelinating regions, mainly motor and sensory systems, which may remain intact in AD until very late stages of the disease [13–16]. Some studies also suggest that AD is a developmental disorder, which can only occur once myelination has been completed [13, 14]. One recent study has also revealed a defect in the biosynthesis of myelin lipids in the preclinical stage of AD, which contributes significantly to the deterioration of the synaptic function and to cognitive decline [16]. What is interesting, the impaired formation of the myelin sheath seems to even precede the pathology of neurofibrillary tangles in AD patients, which indicates that myelin damage may be the first neuropathological abnormality in AD [16]. The above data suggest that AD may be a demyelination disorder. Over time, myelin damage may contribute to synaptic dysfunction and cognitive decline. The article presents current knowledge on the role of myelin in AD, as well as its interactions with amyloid depositions in the brain of AD patients.
Data on the role of myelin from animal models
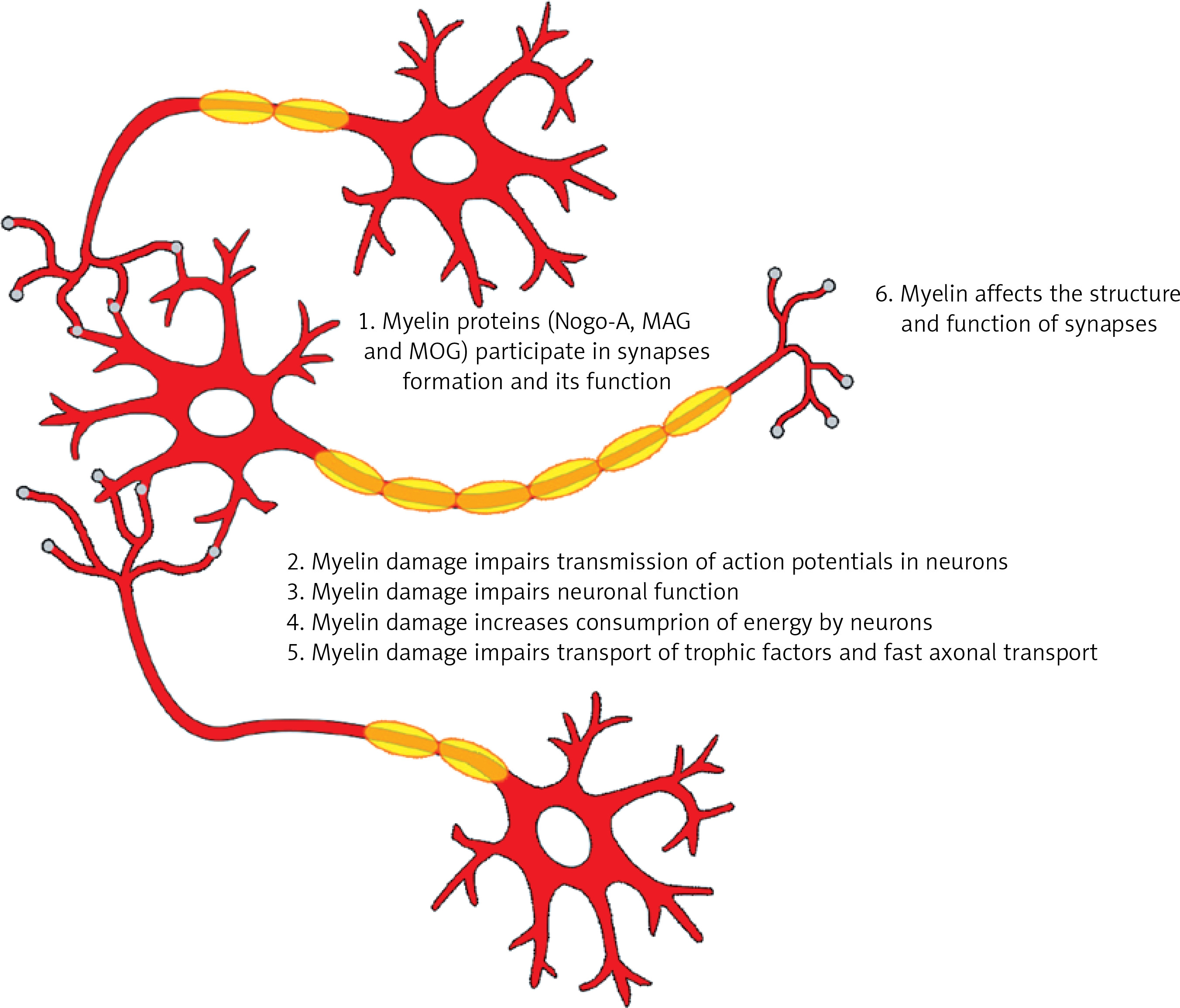
Myelin is wrapped around the axons of neurons and forms structures called nodes of Ranvier where the action potentials are generated, to be then propagated along the myelinated axons [17]. The conduction velocity along the myelinated axons not only depends on axonal diameter, but is also regulated by the number and geometry of the nodes of Ranvier [17]. Myelin impairments may affect transmission of action potentials in neurons. Data from neuropathological animal studies revealed decreased synaptic staining and impaired synaptic transmission in the demyelinated hippocampus of mice with experimental autoimmune encephalitis (EAE) [18, 19]. This finding confirms that myelin plays an important role in synaptic transmission.
Additionally, proteins such as neurite outgrowth inhibitor-A (Nogo-A), myelin-associated glycoprotein (MAG), and oligodendrocyte myelin glycoprotein (MOG), which are expressed in oligodendrocytes and myelin, act as inhibitors of axonal sprouting, which is important for the formation of synapses [20]. But in addition to these proteins, many other factors may contribute to an impaired structure and function of synapses. As a result of demyelination, dysfunction of axonal transport may occur. Proper myelin function also has a beneficial influence on neuronal function. Myelin impairment may impair neuronal function, as myelin supports axonal survival. Oligodendrocytes have the capacity to secrete trophic factors such as insulin-like growth factor-1 (IGF-1), glial cell-derived neurotrophic factor (GDNF), and nerve growth factor (NGF), which exert beneficial effects on neuronal survival [21–23]. Clinically this translates into deterioration of cognition, as the different myelination disorders result in cognitive decline.
Data from experimental animal models of AD also suggest that focal demyelination can be mainly found in the proximity of Aβ plaques within the neocortex [10, 11, 24]. Schmued et al. found evidence for complete disruption of myelinated fibers passing through Aβ plaques or in the proximity of the plaques in the hippocampal region in rats [24]. Thus, various data from animal models show that demyelination may cause neuronal and axonal degeneration. Both of these factors may underlie the cognitive impairment observed in AD.
Data from animal models also provide evidence for the interaction between tau pathology and myelin impairment. Myelin damage has been found to occur prior to the deposition of Aβ plaques and neurofibrillary tangles in the brains of murine models of AD [25, 26]. A defect in myelin biosynthesis has been found in AD subjects even in very early, preclinical stages of the disease, e.g. Braak stage I/II within the temporal cortex, which also confirms that myelin dysfunction precedes amyloid pathology [16]. Couttas et al. observed a significant reduction in the activity of ceramide synthase 2, which is responsible for synthesis of very long chain ceramides belonging to the lipids of myelin sheath. This finding may support the hypothesis that demyelination may have a driving influence in AD pathogenesis [16].
Myelin impairment is probably an early phenomenon in AD pathology, which precedes the onset of typical pathological changes such as Aβ plaques and neurofibrillary tangles. What is interesting, there is also evidence that tau protein hyperphosphorylation may occur later, during the remyelination process [27]. It should be noted here that hyperphosphorylated tau protein, as a marker of axonal and neuronal loss, has also been found in other demyelinating disorders [28]. It is still unclear whether myelin damage really triggers the occurrence of AD pathology.
The role of myelin in cognition
The development of myelin progresses throughout childhood and adolescence, until adulthood [29]. Myelination of the central nervous system is completed at different time points in different parts of the brain. In humans, the development of myelin in the corpus callosum, unlike the formation of neurons, is not completed until the second decade of life [30], and in the frontal lobes, it is not completed until the age of about forty [31]. Myelination is directly connected with the development of cognition and motor function.
Data from animal models show that expression of the myelin basic protein (MBP), which is an important compound of the myelin sheath, is significantly higher in rats which have received training in learning tasks than in rats which have not received such training, and the MBP level is directly associated with the learning rate [32, 33]. What is interesting, in adult mice which had had to learn complex motor skills, the occurrence of new oligodendrocytes and myelin was found within the corpus callosum [34, 35].
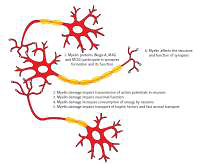
The integrity of the myelin sheath decreases with age, and so does the amount of myelin in the brain. The myelin content reaches a peak in middle age and gradually declines in later years [14, 36]. There is evidence that the age-related cognitive decline is associated with changes in the white matter [37], which may be caused by marked demyelination and the loss of oligodendrocytes. Age-related changes in the integrity of the white matter are associated with cognitive decline in healthy elderly persons [38]. A point worth noting is that there is evidence that memory training can increase the integrity of the white matter in elderly people and at the same time improve their cognitive capacity [39]. To conclude, myelin damage connected with aging may contribute to the cognitive decline in healthy elderly persons (Figure 1).
Figure 1
The role of myelin in cognition. The figure presents the results of myelin damage for cognition. Myelin is responsible for transmission of action potentials. Myelin proteins participate in formation of synapses and their proper function. Myelin damage impairs neuronal function, increases consumption of energy by neurons and impairs transportation of trophic factors as well as fast axonal transport
Myelin damage in AD
The amyloid hypothesis has to be verified, as the correlation between amyloid plaque lesion load and symptoms of AD is very poor [31]. There is also only a weak correlation between amyloid burden in the brain of AD patients and the intensity of brain atrophy [40]. Additionally, amyloid plaque removal treatments failed in terms of clinical improvement in AD patients, although this treatment turned out to be successful in decreasing brain amyloid burden [41, 42]. Soluble oligomeric forms of Aβ can cause synaptic loss, even earlier, before their deposition into neuritic plaques [43]. This phenomenon may impair synaptic transmission, eventually leading to memory impairment [43]. There is multiple evidence that synaptic number is the best correlate of cognition in AD [44]. Recent data suggest that Aβ deposition is important in AD pathology, but it follows rather than precedes myelin damage, which also impairs synaptic transmission. One should not discount the contribution of Aβ to AD pathology, but rather highlight the role of other mechanisms that trigger Aβ and tau production, oligomerization and deposition.
Although there exists evidence for myelin damage in the normal aging brain [9, 13, 31], recent data suggest that AD pathology is influenced by myelin damage [9]. Also neuroimaging studies of patients with MCI reveal white matter damage and myelin impairment in AD patients, before the fifth decade of their lives [45]. The neurodegenerative process in AD was previously perceived as being initiated by the accumulation of aggregated Aβ 42 and the presence of neurofibrillary tangles. This process probably causes the death of neurons. However, there is evidence that it may also cause damage of myelin and myelin-producing oligodendrocytes [14, 46]. Nevertheless, the exact nature of the interaction between AD pathology and myelin damage is still unclear.
Our knowledge on the role of myelin in neuropathology of AD is mainly limited to MRI neuroimaging studies [9, 13, 14, 47, 48]. The most recent MRI data show that beta amyloid deposition in the brain may change the white matter microstructure even in early stages of the disease [9]. Dean et al. described white matter changes within the myelin, especially in late-myelinating brain regions, in a group of 71 subjects with preclinical AD [9]. The authors also suggested that amyloid pathology probably influences the myelin microstructure, and that myelin damage may even precede amyloid deposition in human brains [9]. In addition, Dean et al. found a strong correlation between a decrease in Aβ levels in the cerebrospinal fluid of subjects in the preclinical phase of AD and a decrease in selected MRI myelin measures indicatory of myelin damage [9]. Data from neuroimaging studies also show that there is a relationship between the decrease in the integrity of the white matter and cognitive decline in AD patients [49].
There are also data from biochemical studies which confirm myelin damage in AD. Some studies suggest that myelin damage in AD may even precede Aβ and tau pathologies [14, 16]. The myelin basic protein (MBP), which is an intracellular protein and a major structural protein component of myelin, has been proven to bind Aβ and inhibit Aβ fibril formation in AD, which possibly may have a regulatory role in the deposition of Aβ 42 and formation of amyloid plaques in the extracellular space of the brains of AD patients [50, 51]. MBP is also responsible for Aβ degradation in vitro [50]. MBP levels are significantly decreased in the white matter of AD patients [52], and there is a strong correlation between decreased MBP levels and the increase in Aβ 42 in the brain tissue of AD patients [9]. It is, therefore, possible that the loss of myelin and the decrease in MBP levels result in accelerated deposition of Aβ and increased deposition of Aβ plaques in the brains of AD patients. On the other hand, as mentioned above, the deposition of Aβ in the human brain worsens the state of myelin. It has been shown that Aβ induces oligodendrocyte death and inhibits the formation of myelin [53]. Thus, loss of myelin in AD may be involved in a kind of vicious circle, which promotes further neuronal loss and disease progression.
The above data are also in accordance with the results of biochemical studies which show that there are increased levels of antibodies against different glial-derived antigens (anti-MOG, anti-MAG, anti-MBP, anti-PLP) in sera of AD patients in comparison to healthy control subjects [54]. The increased levels of antibodies against different antibodies of the myelin sheath are probably secondary to myelin damage in AD patients. Of course, it is still unclear whether the presence of these antibodies reflects a diffuse CNS injury or contribute to this injury. Nevertheless, the process of myelin damage probably leads to the presentation of new antigens to the immune system, and subsequent activation of T and B cells.
Also post mortem studies of human brains with AD pathology have revealed a decrease in the number of oligodendrocytes within the sensory motor cortex, superior temporal gyrus, and middle frontal gyrus [55]. Data from autopsy studies indicate that the levels of myelin-associated proteins and myelin lipids are decreased in the early stages of AD, as well as in mild cognitive impairment (MCI) [52, 55].
The cause of myelin damage in AD
Aging is an obvious risk factor for myelin damage, as well as for AD. The number of oligodendrocytes decreases with age by 27% from adolescence until the age of 90 and the volume of the white matter also decreases by 28% within this time. Thus the rate of oligodendrocyte death and myelin damage is (on average 9.5%) faster than the rate of neuronal death [9, 56, 57]. The aging of oligodendrocytes leads to a decrease in the quality and amount of myelin.
Oligodendrocytes originate from oligodendrocyte progenitor cells (OPCs), which have the capacity to clear Aβ through the process of phagocytosis and autophagy [58]. In AD, the number of OPCs and oligodendrocytes becomes smaller, which hinders Aβ cleavage from the brain. As mentioned above, oligodendrocytes are also involved in the neurodegenerative process, which also decreases their number in the course of AD.
Also the inflammatory process in AD may contribute to the myelin damage. Microglia activation and release of inflammatory cytokines may also underlie the myelin damage. Microglial activation also plays an important role in the removal of toxins from the brain, which is crucial for re-myelination. Activation of microglia constitutes a response to the deposition of Aβ in the brain, as it decreases the amount of neurotoxic soluble Aβ in the brain [59]. Additionally, the process of microglial activation is also involved in the clearance of re-myelination inhibitors from the brain, thus promoting the process of creation of new myelin sheaths on demyelinated axons [59]. Microglia in AD subjects, however, exhibit a decreased capacity to clear toxic and harmful substances from the brain, which also contributes to myelin damage [60].
It is worth noting that there exists a relationship between the presence of the ApoE4 allele and the level of myelin damage in AD. ApoE, a confirmed risk factor for the disease, plays an important role in the transportation of endogenously produced brain lipids and recycling of these lipids, which is crucial for the production of myelin, its maintenance, and its repair [31, 61, 62]. It has been proven that apoE4 allele carriers have lower levels of ApoE molecules in serum and the brain tissue than non-carriers [62]. The Apo E4 allele decreases the formation of myelin in the human brain and promotes age-related myelin damage [14].
The similarity of myelin damage in multiple sclerosis and AD
Multiple sclerosis (MS) is one of the most frequent demyelinating disorders, which, like AD, is accompanied by impaired cognition [63]. Cognitive impairment affects 43–70% of MS patients [64]. Multiple data from neuropathological and neuroimaging studies confirm myelin damage in the brains of MS patients [64–67]. It is also known that myelin damage in MS contributes to cognitive decline [68, 69]. What is more interesting, there exists evidence that spatial memory impairment and working memory impairment are especially connected with demyelination of the CNS [70]. The data from MS research suggest that myelin damage may directly lead to cognitive decline and memory deficits.
Myelin damage is also typical of the normal aging brain, and research shows that it is this damage that contributes to the cognitive decline in elderly people [71]. Age-related cognitive decline is mainly due to marked demyelination and loss of oligodendrocytes [71, 72]. Additionally, there is evidence that memory training may increase the integrity of the white matter and improve memory both in MS patients [64] and elderly people [39].
Both MS and AD are accompanied by an inflammatory process and both are neurodegenerative disorders. Inflammation may contribute to myelin damage. Both activated microglia and inflammatory cytokines may be involved in myelin impairment. Demyelination may, in turn, contribute to the dysfunction of neurons, which forms a kind of vicious circle both in AD and in MS, and may promote further progression of these diseases.
Conclusions
There is accumulating evidence that myelin impairment is an important part of the pathological changes observed in AD. Data from most recent studies suggest that Aβ and tau proteins may be by-products of myelin repair in AD rather than being the main underlying cause of dementia [9, 31]. These data are also supported by the fact that previous attempts to control the clinical symptoms of AD by removing Aβ from the human brain failed, even though different agents turned out to be successful in Aβ removal from the brain tissue. At present, it is difficult to say whether myelin damage itself is sufficient to drive the neurodegenerative process and the cognitive decline. We are also lacking strong evidence to answer the question why myelin damage may lead to increased Aβ levels and tau hyperphosphorylation.
Although AD pathology used to be typically linked with neuronal degeneration, most recent data show that it is strongly associated with pathology of oligodendrocytes and myelin [9, 31, 54]. These data are supported by human MRI neuroimaging studies, but also by biochemical and post mortem studies mentioned above. Recent findings suggest that myelin probably plays a more important role in AD pathology than previously thought, but further biochemical, radiological, and neuropathological studies are necessary to discover the real role of myelin in AD pathology.