Introduction
Hypertrophic cardiomyopathy (HCM) is an inherited cardiac disorder, defined by the presence of increased left ventricular (LV) wall thickness, that is not solely explained by abnormal loading conditions [1–4]. In the majority of cases, HCM is inherited as an autosomal dominant genetic trait with a 50% risk of transmission to offspring [5]. It is the most frequently inherited heart disease, with prevalence estimated as between 1 in 200 and 1 in 500 individuals [6], and is the most common cause of sudden cardiac death in young athletes [7], who are often unaware of their underlying condition. Genetic screening is an important tool for the clinical management of HCM patients and their families. Although specific genotype-based treatments for HCM are not yet available, positive genetic test results confirm the etiology of the disease and enable mutation-specific confirmatory testing of the appropriate family members.
Nevertheless, the yield of genetic testing is very variable. The sequencing of sarcomere protein genes identifies a disease-causing mutation in 20–60% of cases [8–10], but different criteria were used to define a variant as disease-associated in these reports. The Mayo HCM Genotype Predictor score (Mayo Score) was published in 2014 for predicting the diagnostic yield of genetic testing, based on clinical and echocardiographic variables [10]. Nine HCM-associated myofilament genes were evaluated in the original cohort of Mayo Score patients [10].
The aim of this study was to assess the yield of genetic testing using next generation sequencing (NGS), evaluating up to 229 genes using the classification according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines, and to validate the previously established and published Mayo Score in the national HCM cohort using these stringent classification criteria.
Material and methods
We included 336 consecutive unrelated patients (41% women, mean age: 53 ±15 years) with a clinical diagnosis of HCM in a single tertiary cardiovascular center in our study. The DNA samples for NGS testing were obtained from 2005 to 2017. Only adult patients were included in our study. Diagnosis was established by experienced cardiologists based on clinical examination, electrocardiography and findings of left ventricular (LV) hypertrophy ≥ 15 mm on echocardiography and/or magnetic resonance imaging [1, 3]. Secondary hypertrophy attributable to aortic valve stenosis or amyloidosis was excluded. When patients presented with mild concomitant systemic hypertension, the HCM echocardiography specialists had to claim the hypertension to be either controlled or the severity insufficient to cause the degree of LV hypertrophy. The echocardiographic septal contour was assessed and categorized as reverse curve, sigmoid, apical or neutral contour according to published criteria [11]. Demographic, clinical and echocardiographic data were collected in all patients. All HCM patients were regularly followed up every 3–12 months according to their current clinical state.
NGS findings were correlated with Mayo Scores [10], based on clinical and echocardiographic variables (septal reverse curve morphology, age at diagnosis < 45 years, maximum left ventricular wall thickness ≥ 20 mm, family history (FH) of HCM, FH of sudden cardiac death, hypertension). The total Mayo Score, ranging from –1 to 5 for each patient, was compared with the yield of genetic testing.
Next generation sequencing
Targeted next-generation sequencing enrichment panel
Genomic DNA was isolated from whole blood using a FlexiGene DNA AGF3000 Kit (QIAGEN, Germany) on the AutoGen Flex 3000 (AutoGen, Holliston, USA) according to the manufacturer’s instructions. Three different gene enrichment panels were used for DNA library preparation according to availability, current knowledge and clinician or laboratory requirements: TruSight Cardiomyopathy (Illumina, San Diego, USA; 46 genes), TruSight Cardio (Illumina, San Diego, USA; 174 genes), and a custom-made SeqCap EZ choice library kit comprising coding regions of 229 cardiac conditions-related genes (Roche NimbleGen, Madison, USA). DNA library preparation was performed according to the manufacturer’s instructions. DNA libraries were sequenced by NGS with paired-end reads (2 × 150 bp cycles) on the MiSeq or MiniSeq platforms (Illumina, San Diego).
Variant identification
We used an “in-house” developed pipeline according to Genome Analysis Toolkit (GATK) best practices for germline variant identification [12, 13]. Raw data from NGS was aligned using the Burrows-Wheeler Alignment Maximal Exact Match (BWA-MEM) algorithm [14] to the human genome reference GRCh37/hg19. PCR duplicates were marked at this step and ignored in the following analyses. Base recalibration was then performed using GATK and variant calling was performed by GATK-HaplotypeCaller [12, 13].
Variant prioritization and classification
We used the Illumina VariantStudio Software v3.0 (Illumina, San Diego) and BaseSpace Variant Interpreter (Illumina, San Diego) for variant prioritization and annotation. Variants with read depth < 10×, quality < 200, synonymous and intronic variants in non-splice regions and minor allele frequency (MAF) higher than expected for HCM [15] were removed for further analysis. We used Sorting Tolerant From Intolerant (SIFT) and PolyPhen-2 prediction tools to assess the potential functional impacts of variants and detect missense variants. We used the National Center for Biotechnology Information (NCBI) ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) and Human Gene Mutation Database (HGMD) databases to assess the clinical impact of variants [16]. Potential variants were classified according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines into one of the following classes: pathogenic, likely pathogenic, uncertain significance, likely benign and benign with respect to HCM [17]. All pathogenic and likely pathogenic variants were validated using DNA Sanger sequencing.
Statistical analysis
Data are presented as means ± standard deviations (SDs) for continuous variables and proportions for categorical variables. The Student’s t test, Mann-Whitney test and Fisher’s exact test were used where appropriate. The level of statistical significance was set to 0.05. The Prism v.7.04 (GraphPad Software Inc., USA) statistical software was used for statistical analysis.
Results
The baseline characteristics of all 336 patients are shown in Table I.
Table I
Patients’ characteristics
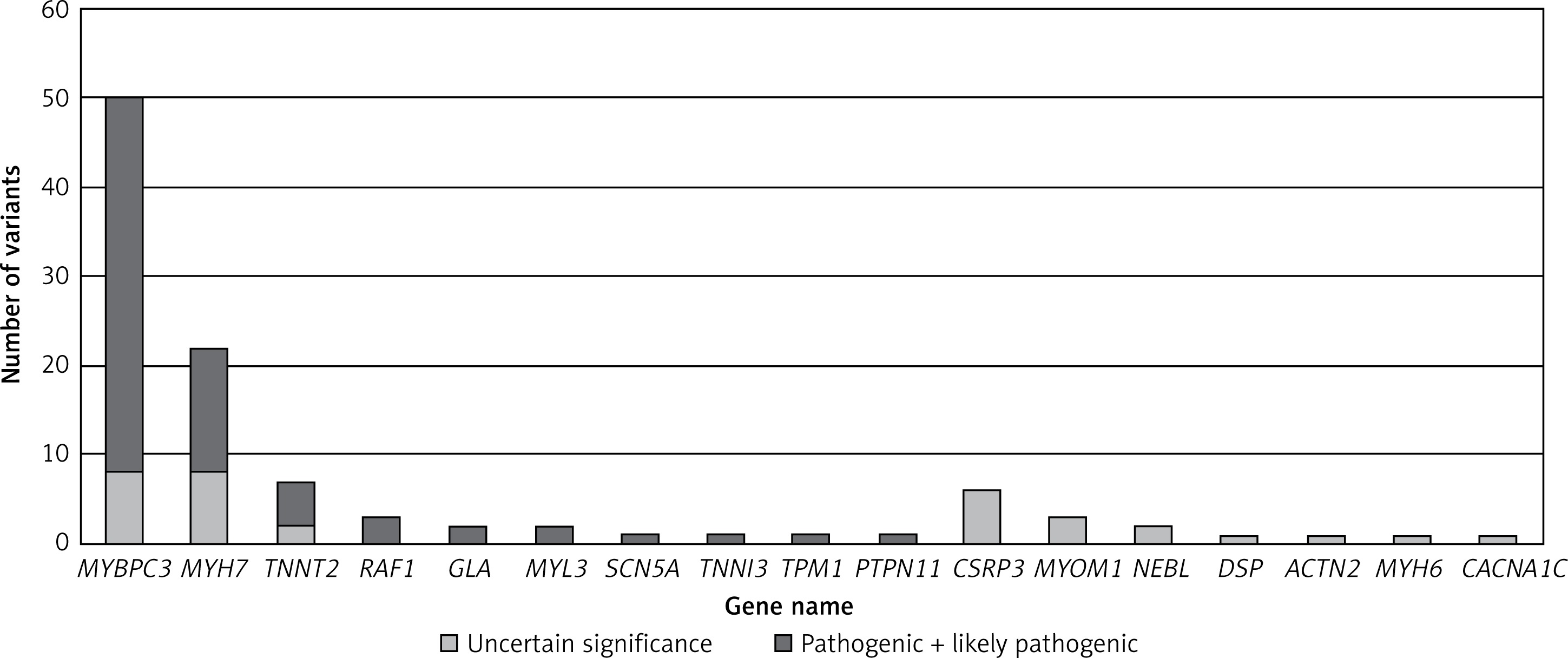
We identified 276 genetic variants in 71 different genes in 188 (56%) HCM patients. All class variants, sorted by the most frequent genes, are shown in Table II. The complete list of all variants in all mutated genes is available in Supplementary Tables SI and SII. Seventy-two variants identified in 70 (21%) HCM patients were classified as pathogenic or likely pathogenic (class 4 and 5 according to ACMG guidelines), as shown in Tables III and IV. A total of 188 variants identified in 142 (42%) HCM patients were classified as pathogenic, likely pathogenic or variants of unknown significance (VUS), as shown in Table V.
Table II
All class variants, sorted by most frequent genes
Table III
Variants’ classification according to ACMG guidelines
Table IV
Variants’ classification according to ACMG guidelines
Table V
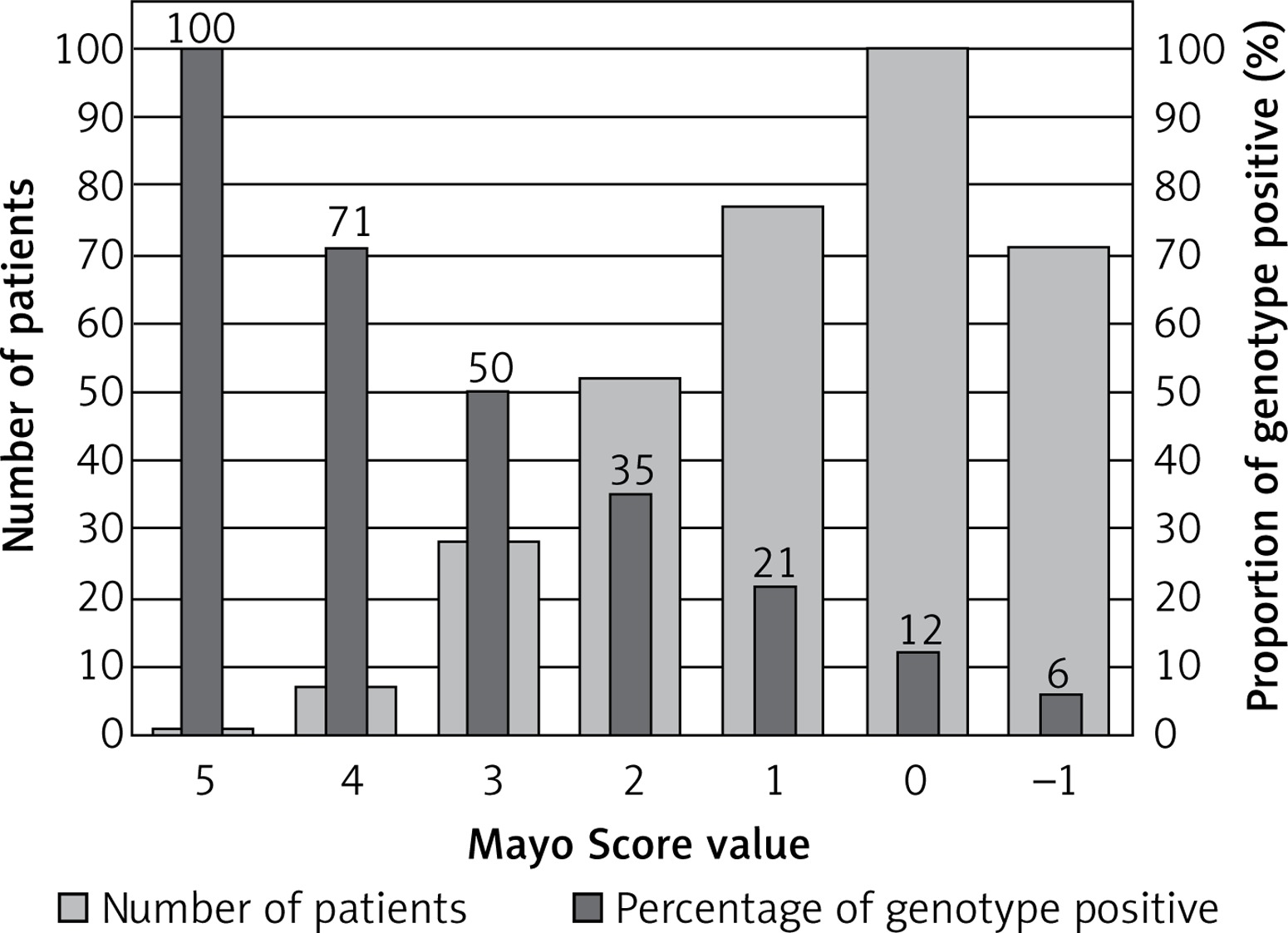
Patients sorted by Mayo Score value and proportion of positive genotype
| Mayo Score | Numbers of patients | Class 4 and 5 (%) | Class 3 to 5 (%) |
|---|---|---|---|
| 5 | 1 | 1 (100) | 1 (100) |
| 4 | 7 | 5 (71) | 5 (71) |
| 3 | 28 | 14 (50) | 16 (57) |
| 2 | 52 | 18 (35) | 29 (56) |
| 1 | 77 | 16 (21) | 39 (50) |
| 0 | 100 | 12 (12) | 31 (31) |
| –1 | 71 | 4 (6) | 21 (30) |
MYBPC3 and MYH7 were two major genes with identified variants, and MYBPC3 (58%) played a dominant role in the group of pathogenic or likely pathogenic genetic variants (Figure 1). A total of 7 pathogenic or likely pathogenic variants were found in the genes RAF1, SCN5A, GLA and PTPN11, as shown in Table III. In the rest of candidate genes, only VUS were identified. No compound heterozygotes of pathogenic or likely pathogenic variants were found. In 38 patients, we identified complex genotypes. The complete list of identified variants is available in Supplementary Table SIII.
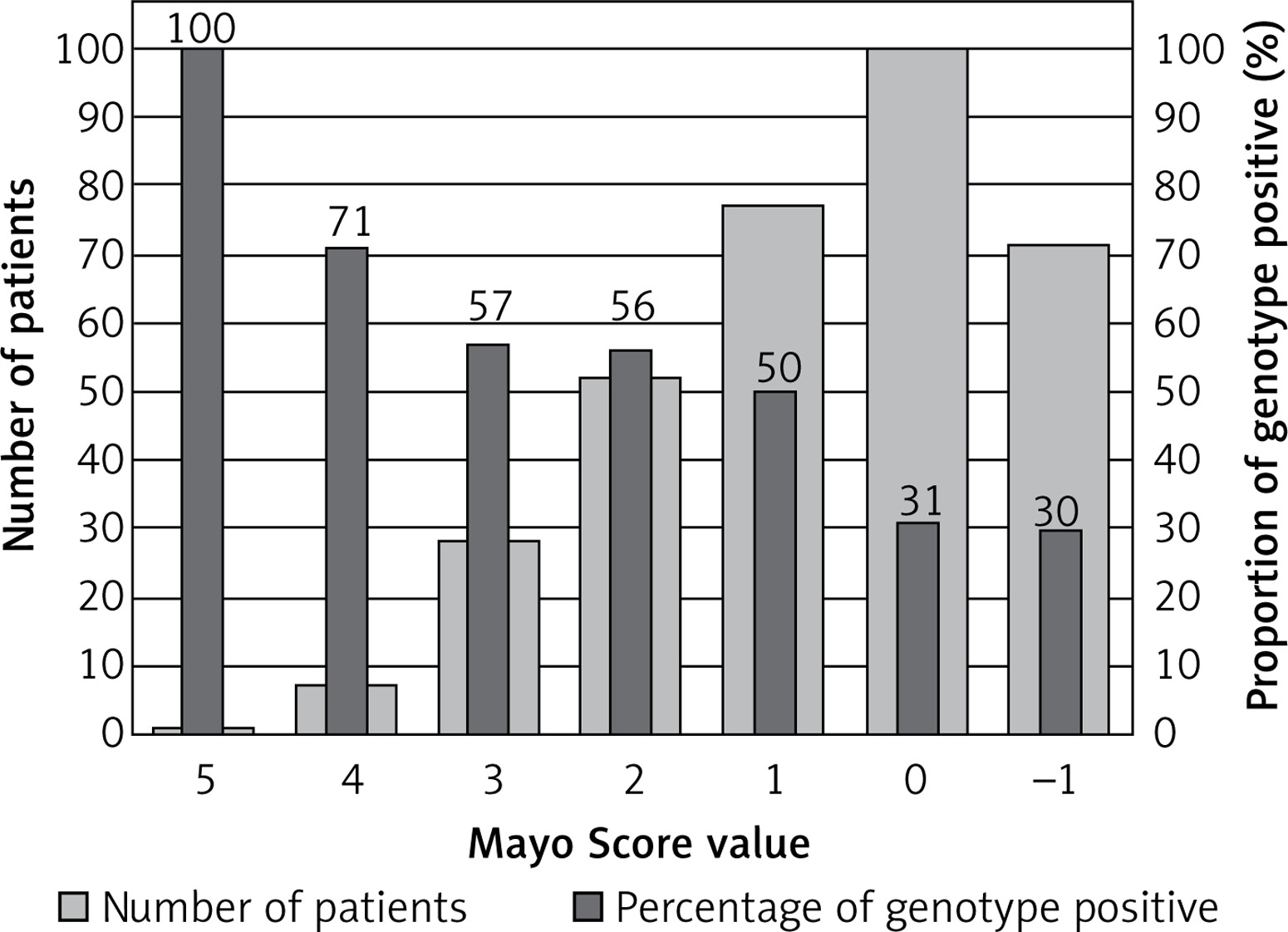
One patient with a Mayo Score of 5 (the highest) had a pathogenic mutation (100% yield). Patients with Mayo Scores of 4 had pathogenic mutations in 71% of cases. Patients with Mayo Scores of 3 or 2 had pathogenic mutations in 50% and 35% of cases, respectively. The yield of genetic testing in patients with a Mayo Score of –1 to 1 was low (6–21%), as shown in Figure 2. Numbers of patients categorized into each Mayo Score value group are presented in Table V. Two analyses of genotype positivity are presented. The Mayo Score values and proportions of positive genotype patients, if all VUS were included, are shown in Figure 3. Cohort demographics and comparison between genotype positive and genotype negative patients are presented in Table VI.
Table VI
Comparison between genotype positive and genotype negative patients
Discussion
Hypertrophic cardiomyopathy is a relatively common genetic cardiovascular disease. Autosomal dominant inheritance is most frequently described [1, 3]. In 1990, a mutation in the β-myosin heavy chain (MYH7) was first identified as responsible for causing HCM [18]. Over the next two decades, numerous genes were reported to be associated with the disease. Nowadays, more than 1500 mutations in 15 or more genes have been described in association with HCM [19–21]. The most vigorous evidence indicates that eight genes are known to definitively cause HCM: MYH7, MYBPC3, TNNI3, TNNT2, TPM1, ACTC1, MYL2, MYL3 [3]. Mutations involving the sarcomere protein genes MYBPC3 and MYH7 are the most common, and phenotypic expression of the disease is heterogeneous, even in families with the same MYBPC3 and MYH7 mutation [19–21]. Our study used massive parallel sequencing and the largest gene panel to date in patients with HCM.
The yield of genetic testing is variable. The likelihood of finding a causal mutation is higher in patients with familial disease and in younger patients [1]. Pathogenic mutations were found in 50–60% of familial HCM and in 30–40% of apparently sporadic cases with no family history [8]. In the original Mayo Score cohort, nine HCM-associated myofilament genes were evaluated (ACTC1, MYBPC3, MYH7, MYL2, MYL3, TNNC1, TNNI3, TNNT2, TPM1) and the overall yield of genetic testing was 34% [10]. Genetic testing gene panels may vary in size depending on the company or institution providing the test. We hypothesized that including a large number of genes could lead to a higher diagnostic yield of genetic testing. This could subsequently enable us to perform cascade mutation-specific confirmatory testing of the appropriate family members more frequently. We therefore evaluated 229 cardiac condition-related genes. All of our three testing gene panels included the main nine genes mentioned.
Genetic testing in unselected patients with a clinical HCM diagnosis leads to practical problems, as current guidelines recommend that all clinically diagnosed HCM patients undergo genetic testing to enable cascade genetic screening of their relatives [1, 3]. If this testing reveals a pathogenic mutation, first degree relatives can undergo mutation-specific confirmatory genetic testing. The genetic and allelic heterogeneity makes molecular analysis by conventional methods time-consuming and expensive [22, 23]. Genomic testing based on NGS allows the rapid analysis of a large number of genes at a similar cost and accuracy to conventional sequencing methods. The genetic heterogeneity of HCM, however, makes genetic screening and interpretation of the results challenging, since the screening of large numbers of genes results in the identification of many genetic variants of unknown significance (VUS) [23, 24].
Our relatively low yield and the low frequency of mutations in certain genes may be a result of the different (and more stringent) criteria used to define a variant as disease-associated over time. In the past, a variant might have been defined as disease-associated based solely on its absence in 50–100 reference alleles in healthy controls [25, 26]. In the original Mayo cohort, case-derived variants that were absent in more than 8400 healthy controls or seen with a frequency of < 0.01% in controls and significantly overrepresented in cases versus controls were included as genotype positive [10]. Two years after the publication of the original Mayo Score paper, however, the same authors used a new cohort of HCM patients to validate the original genotype predictor score [27]. Variants classified as pathogenic, probably or possibly pathogenic, or VUS were considered genotype positive in the validation study [27]. All variants were re-analyzed by determining minor allele frequency (MAF) in population databases such as Exome Aggregation Consortium (ExAC), by reference in clinical databases (ClinVAR) and by the application of various in silico mutation prediction tools (PolyPhen, SIFT, Mutation Assessor, Condel and Grantham score). Variants that were seen at MAF > 0.01% were reclassified as VUS, likely benign, after which the genotype predictor score was recalculated. As a result, the yield of genetic testing for patients with low genotype scores (–1, 0, 1, 2) decreased to 16, 20, 26, and 61%, respectively [27].
All variants in our study were classified according to the ACMG and the AMP guidelines into one of the following classes: pathogenic, likely pathogenic, variants of uncertain significance, likely benign and benign with respect to HCM [17]. Only variants classified as pathogenic and likely pathogenic (class 4 and 5) were considered genotype positive in our study. The most significant difference between the Mayo cohort and our study was that allele frequencies higher than expected for HCM (population frequency < 4 × 10–5) [15] were removed from further analysis in our study. This could explain why the yield of our genetic testing is lower than in Mayo patients (51% in the validation cohort and 34% in the original cohort). If we had used a similar approach in our study, the yield of genetic testing would be higher, but the interpretation of positive genotypes would be less reproducible in future research using the most recent classification guidelines [17].
Only 11% of our patients had FH of HCM, while the reported prevalence was about 30% in most papers [10, 20, 27, 28]. Furthermore, the mean age of our HCM patients is relatively high. When compared to the reported results of similar studies [5, 10, 20, 27], our patients were on average some 8–9 years older at the time of diagnosis. The lower age at diagnosis could correspond with the FH of HCM [1, 29, 30]. In patients with a milder HCM phenotype characterized by late-onset disease, sigmoidal septal shape and concomitant arterial hypertension, the LV hypertrophy might not be primarily genetically caused by sarcomeric protein mutation, but might be a result of age, comorbidities (e.g. diabetes, renal insufficiency), genetic modifiers or environmental risk factors. In older patients with LV hypertrophy and arterial hypertension, coexistence of HCM is often a consideration. As shown in Table VI, the possible predictors of positive genotype are lower age, FH of HCM, reverse curve septal shape and presence of obstruction, whereas arterial hypertension and sigmoid septal shape could predict negative genotype.
Using the Mayo Score in the original study, the likelihood of a positive genetic test ranged from 6% when only hypertension was present (–1 point), to 80% when all five positive predictors were present and hypertension was absent (5 points) [10]. When applied to our cohort, the yield of genetic testing in patients with Mayo Scores of –1 point was exactly the same – 6%. The patient with the highest Mayo Score of 5 had a pathogenic mutation, and therefore we could claim a “100% yield”, but naturally more patients would be needed to prove this. When differences in the prevalence of HCM FH and AH history are considered, low numbers of patients with the highest Mayo Scores are not unexpected. Together with the specific proportion of mutated genes, this could suggest that our national cohort has a unique genetic background and risk profile. Our patients with the second highest Mayo Score (4 points) had pathogenic mutations in 71% of cases, and the rest of our groups, sorted by Mayo Score values, were also comparable with the original cohort [10].
Our hypothesis, that including a large number of genes could lead to a higher diagnostic yield in genetic testing, has not been supported. Despite the fact that 229 cardiac condition-related genes were evaluated, the diagnostic yield remained low when stringent ACMG and AMP guidelines were applied. The pathogenic or likely pathogenic variants were found in only 10 genes. In patients with ventricular arrhythmias, no significant variants in ion channels genes were found, with one exception (patient R.D. with the complex genotype including SCN2B gene class 3 variant, reported in Supplementary Table SIII). Our approach using a wide gene panel might therefore not be applicable to routine clinical practice due to the cost-benefit ratio. Nevertheless, we managed to identify in detail the genetic background of a unique Central European cohort of HCM patients. Some patients would not have been properly diagnosed without our genetic testing. Finding pathogenic mutations in RAF1 and PTPN11 genes helped to confirm the diagnosis in patients with Noonan syndrome and LEOPARD syndrome, respectively. Finding a pathogenic mutation in the GLA gene enabled the early diagnosis of two patients with Fabry disease, which was not clinically apparent at the time of genetic testing. This is particularly clinically important because enzyme replacement therapy might be more effective if given at earlier stages of the disease before the development of LV hypertrophy and myocardial fibrosis [31]. We have also identified a large number of VUS. If archived in clinical databases (e.g. ClinVAR) and if segregation analysis is planned in related patients, it could lead to the discovery of new candidate genes and mutations (e.g. cardiac Z-disk protein nebulette [32], found in two of our patients), reveal new pathophysiological pathways [33] and raise the diagnostic yield in HCM patients in the future. If all patients with VUS were considered to be genotype positive, the diagnostic yield would rise to 42% and still would be predicted with Mayo Score values, as presented in Table V and Figure 3.
Our study has several limitations. It was a single center study. Hence we cannot be confident that these data are entirely representative and generalizable for non-tertiary referral centers with less experience in the management of HCM patients. Secondly, three different gene enrichment panels were used for DNA library preparation according to availability, current knowledge and clinician requirements. Nevertheless, 82% of all patients’ samples were evaluated with the largest gene panel within the years 2015–2017. Thirdly, the study was performed using the national HCM cohort, so the results might not be fully applicable except to other Central European patients. Fourthly, pediatric patients were not included in our study. Therefore, the results are only applicable to the adult HCM population.
In conclusion, the overall yield of genetic testing using NGS to evaluate up to 229 genes was low. A large number of genetic variants in multiple genes, and the dominant role of the MYBPC3 gene, were identified. Only one fifth of variants were classified as pathogenic or likely pathogenic, however, when stringent ACMG and AMP guidelines were applied. The yield of genetic testing was consistently predicted with Mayo Score values.