

Introduction
Periodontitis is one of the most common chronic inflammatory diseases leading to the destruction of connective tissue and alveolar bone surrounding the teeth [1, 2]. Porphyromonas gingivalis(Pg) is the main pathogen to initiate this disease, and host immune responses are critical to the progression of periodontitis [3, 4]. The initial periodontitis is characterized by infiltration of inflammatory cells such as leukocytes, macrophages, and lymphocytes.
Macrophage activation is an indispensable process in innate immunity. Macrophages located in the oral mucosa produce pro-inflammatory cytokines such as tumor necrosis factor α (TNF-α), interleukin (IL)-1β, IL-6, IL-12, IL-17, and monocyte chemotactic protein-1 (MCP-1); these macrophages responding to Pg were classified into the M1 pro-inflammatory macrophage subset [5-8]. M1 macrophages upregulate the expression of inducible nitric oxide synthase (iNOS), which subsequently results in tissue damage [5, 7, 9, 10]. M1 macrophages, which are considered as osteoclast precursors, produce cytokines including IL-1β, IL-6, and TNF-α that promote alveolar bone resorption, and proteases such as metalloproteinase and collagenase, which further damage periodontal connective tissue [11, 12].
Uncoupling proteins (UCPs) are located in the mitochondrial inner membrane; they transport protons from the intermembrane space of mitochondria into the matrix and reduce the mitochondrial membrane potential. UCP2 is the most wildly expressed homologue among UCPs. UCP2 plays an essential role in regulating mitochondrial ATP generation, maintaining calcium homeostasis, and reactive oxygen species (ROS) elimination [13].
Evidences indicate a critical role of UCP2 in macrophage activation. For example, UCP2-deficient macrophages promotes more inflammatory cytokines and ROS production [13-16]. In addition, lipopolysaccharide (LPS)-treated UCP2 knockout (UCP2-KO) mice had enhanced NF-kB activation due to increased ROS production [15], overexpression of UCP2 in macrophages could decrease ROS production and inhibit macrophage activation [14], and increased NO and cytokine production by macrophages was found in UCP2-KO mice with autoimmune diabetes [17].Considering the indispensable role of macrophages in the development of periodontitis and the essential role of UCP2 in macrophage activation, we hypothesize that UCP2 may represent a critical role in the pathogenesis of periodontitis.
In the present study, we used Porphyromonas gingivalis lipopolysaccharide (Pg-LPS) to induce periodontitis in mice and applied macrophage-specific UCP2 knockout mice to investigate the role of UCP2 in macrophage activation in periodontitis. Our results suggest that UCP2 is a pro-pathogenic mediator in periodontitis, whereby UCP2 regulates ROS production as well as proliferation and migration ability of macrophages.
Material and methods
Collection of human gingival samples
Gingival tissues were obtained at the Department of Stomatology in Jinling Hospital. The experimental protocols was approved by the Ethics Committee of Jinling Hospital (registration number: 2014GJJ-114). Gingival tissues were collected from 10 periodontitis patients, aged 39-52 years. Patients diagnosed with chronic periodontitis (CP) met the Armitage diagnostic criteria [18, 19]. At least two loci from patients with severe CP were selected, having pocket depth (PD) > 6 mm, clinical attachment level (CAL) > 5mm, tooth mobility (3 degree), and bleeding on probing (+). The control group was selected from the same patients with healthy gingiva (plaque index [PI] = 0, gingival index [GI] = 0, and PD ≤ 3 mm). The excluding criteria were as follows: smoking or history of smoking, systemic disease, antibiotics or mouthwash within 6 months, periodontal disease treated within 6 months.
The way to obtain periodontitis tissue included clinically tested subject for the degree of tooth looseness III, and the periodontal pocket detected over 6 mm. The clinical symptoms were diagnosed as the affected tooth, which could not be retained in severe periodontitis. In order to achieve the goal of complete debridement of the alveolar socket for later implantation, the teeth were removed to obtain the infected periodontal tissue around the affected tooth.
The way to obtain self-control health gingiva: In the experimental group, after three months of tooth extraction, dental implant repair was performed and after the incision of gingival tissue, a portion of healthy gingival tissue was obtained with the patient’s informed consent.
Half of the excised samples were used for RNA extraction, while the other half were used for immunochemical examination.
Animals
UCP2fl/flmice (B6; 129S-Ucp2tm2.1Lowl/J, 022394) and csf1r-iCre mice (FVB-Tg; Csf1r-cre/Esr1*.1Jwp/J, 019098) were obtained from the Jackson Lab and maintained according to the guidelines of the Institutional Animal Care and Use Committee at Nanjing Medical University (permission number: SYXK2016-0016). Macrophage-specific UCP2 knockout mice were generated by crossing UCP2fl/flmice and csf1r-iCre mice. UCP2fl/fl, iCre+/– mice were used as MφUCP2–/– mice, and their littermates with genotype UCP2fl/fl, iCre–/– were considered as MφUCP2+/+ mice. UCP2fl/fl, iCre+/– mice and their littermate control mice were intraperitoneally injected with tamoxifen (cat: T5648, Sigma-Aldrich, USA) at 25 mg/kg for 5 consecutive days before Pg-LPS injection.
Pg-LPS (cat: tlrl-pglps, InvivoGen, USA) (25 µl in 3 µl PBS) or control (3 µl PBS only) was injected every 48 hours into the mesial gingiva of left mandible of mice under isoflurane anesthesia according to previous studies [20, 21]. Mice were euthanized 28 days after injection.
Cell culture and treatment
Bone marrow-derived macrophages (BMMs) were isolated from the bone marrow, as previously reported. To generate UCP2–/– macrophages, BMMs isolated from MφUCP2–/– mice and littermate control mice were cultured in RPMI-1640, containing 10% fetal bovine serum (Invitrogen, Grand Island, USA), 1 µM 4-hydroxytamoxifen (H6278, Sigma-Aldrich), and 10 ng/ml mouse m-CSF (cat: 416-MI-050; R&D, USA). On the 7th day of culture, BMMs were cultured with serum-free medium and treated with Pg-LPS (100 ng/ml) for 24 hours.
Western immunoblot analysis
Cells were lysed in RIPA buffer (Beyotime, China) supplemented with 100 mg/ml PMSF, 1% protease inhibitor cocktail, and 1% phosphatase II and III inhibitor cocktail (Sigma-Aldrich, USA). Protein concentration was determined by bicinchoninic acid protein assay. An equal amount of protein was loaded into a 10% SDS-PAGE gel and transferred into polyvinylidene difluoride membranes. The primary antibodies used were as follows: anti-iNOS (ab-178945; Abcam, Cambridge, UK), anti-UCP2 (sc-6527; Santa Cruz Biotechnology, USA), and anti-β-actin (ab-8226; Abcam, Cambridge, UK). Western blots were performed at least three times independently. Quantification was performed by measuring the intensity of signals with the aid of the National Institutes of Health (NIH) ImageJ software package.
PCR
RNA extraction and cDNA synthesis were performed as previously described. Briefly, total RNA was prepared using TRIzol reagent (invitrogen) according to the manufacturer’s instructions. The first strand of cDNA was synthesized using 1 µg of total RNA, Rever Tra Ace (Vazyme, China), and oligo (dT) 12-18 primers. Gene expression was measured by real-time PCR, using real-time PCR Master Mix reagents (Vazyme, China) and 7300 real-time PCR system (Applied Biosystems, Foster City, CA, USA). GAPDH was used as an internal control. The relative amount of mRNA was calculated using an equation 2-ΔCT, in which ΔCT = CT gene – CT control.
Measurement of macrophage proliferation activity
BMMs were isolated and cultured in 6-well plates with RPMI-1640 containing 10% FBS, M-CSF (10 ng/ml), and 1 µM 4-hydroxytamoxifen. On the 3th day of culture, macrophages were seeded in 96-well plates at 1 × 104 cells per well and cultured with RPMI-1640 containing 10% FBS, M-CSF, and 4-hydroxytamoxifen. After 4 days of culture, BMMs were cultured with serum-free medium and treated with M-CSF (10 ng/ml) for 24 hours. Ten microliter of cell counting kit-8 (CCK-8) solution (Beyotime, China) was added to the each well in the absence of light and incubated at 37°C for 4 hours. The absorbance of each well at 450 nm was determined using an enzyme-linked immunosorbent assay reader.
Histology assay
Briefly, the upper jaws of all mice were decalcified using 14% EDTA in PBS for 3-4 weeks, and the samples were then embedded in paraffin and sectioned at 5 mm thickness for hematoxylin and eosin (H&E) staining, and toluidine blue staining. Slides were visualized with a Nikon Eclipse 80i microscope equipped with a digital camera (DS-Ri1; Nikon, Shanghai, China).
Immunofluorescent staining
Gingival cryosections at 3 mm thickness were fixed for 15 min in 4% paraformaldehyde, and then permeabilized with 0.2% Triton X-100 in PBS for 5 min at room temperature. After blocking with 2% donkey serum for 60 min, the slides were immunostained with primary antibodies against F4/80 (no. 14-4801; eBioscience, San Diego, USA), iNOS (ab-178945; Abcam, Cambridge, UK), or UCP2 (sc-6527; Santa Cruz Biotechnology, USA), followed by staining with FITC/TRITC-conjugated secondary antibody. The slides were double-stained with DAPI to visualize the nuclei and viewed with a Nikon Eclipse 80i Epi-fluorescence microscope equipped with a digital camera (DS-Ri1, Nikon, Shanghai, China).
Migration assays
UCP2–/– macrophages and UCP2+/+ macrophages were cultured in 12-well plates and treated with Pg-LPS (100 ng/ml). The culture plates were scratched by a sterile micropipette tip, and images were taken at 0, 24, and 48 hours after scratching. Migration rates were calculated based on the relative changes of the gap.
Transwell cell migration assay was performed according to a previous report. Briefly, BMMs at 1 × 104 cells were seeded in the upper compartment of 8 µm pore transwell chambers (cat: PIEP12R48, Millipore, USA), and Pg-LPS was added to the lower compartment. After 24 hours of incubation at 37°C in 5% CO2, the upper chamber was fixed with methanol and stained with 0.5% crystal violet (Beyotime, China) for 20 min. The stained cells were subsequently photographed. Next, the stained cells were further decolorized by 33% acetic acid, and the OD value at 570 nm was measured.
Measurement of intracellular reactive oxygen species
Reactive oxygen species (ROS) generation was estimated by using a reactive oxygen species assay kit (Beyotime, China). After treatment with Pg-LPS for 24 hours, macrophages were incubated with 10 µmol/l 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA), incubated at 37°C for 20 min, and then observed by flow cytometry.
Measurement of NO levels
The levels of NO in the culture medium and macrophage were estimated by using a nitric oxide assay kit Griess reagent according to the manufacturer’s instructions (Beyotime, China).
Flow cytometry
Approximately 1 × 106 cells were stained for 20 min with antibodies including anti-CD45-APC (561018; BD Biosciences, USA), anti-CD86-FITC (130-102-506; Miltenyi Biotec, Germany), and anti-F4/80-PE (12-4801-80; eBioscience, USA), washed, and resuspended in FACS buffer. The cell suspensions were then washed twice with FACS buffer, resuspended in FACS buffer, and analyzed on BD Canto II Flow Cytometer with FlowJo software.
Detection of cytokine and chemokine
The concentration of regulated upon activation normal T cell expressed and secreted factor (RANTES), MCP-1, IL-1β, and IL-6 in gingiva was measured using the Milliplex Map assay (cat: Merck Millipore, Shanghai, China) via Luminex 200 system (Luminex Corporation, Texas, USA).
Results
UCP2 was upregulated in the macrophages infiltrating human periodontal tissues affected with periodontitis
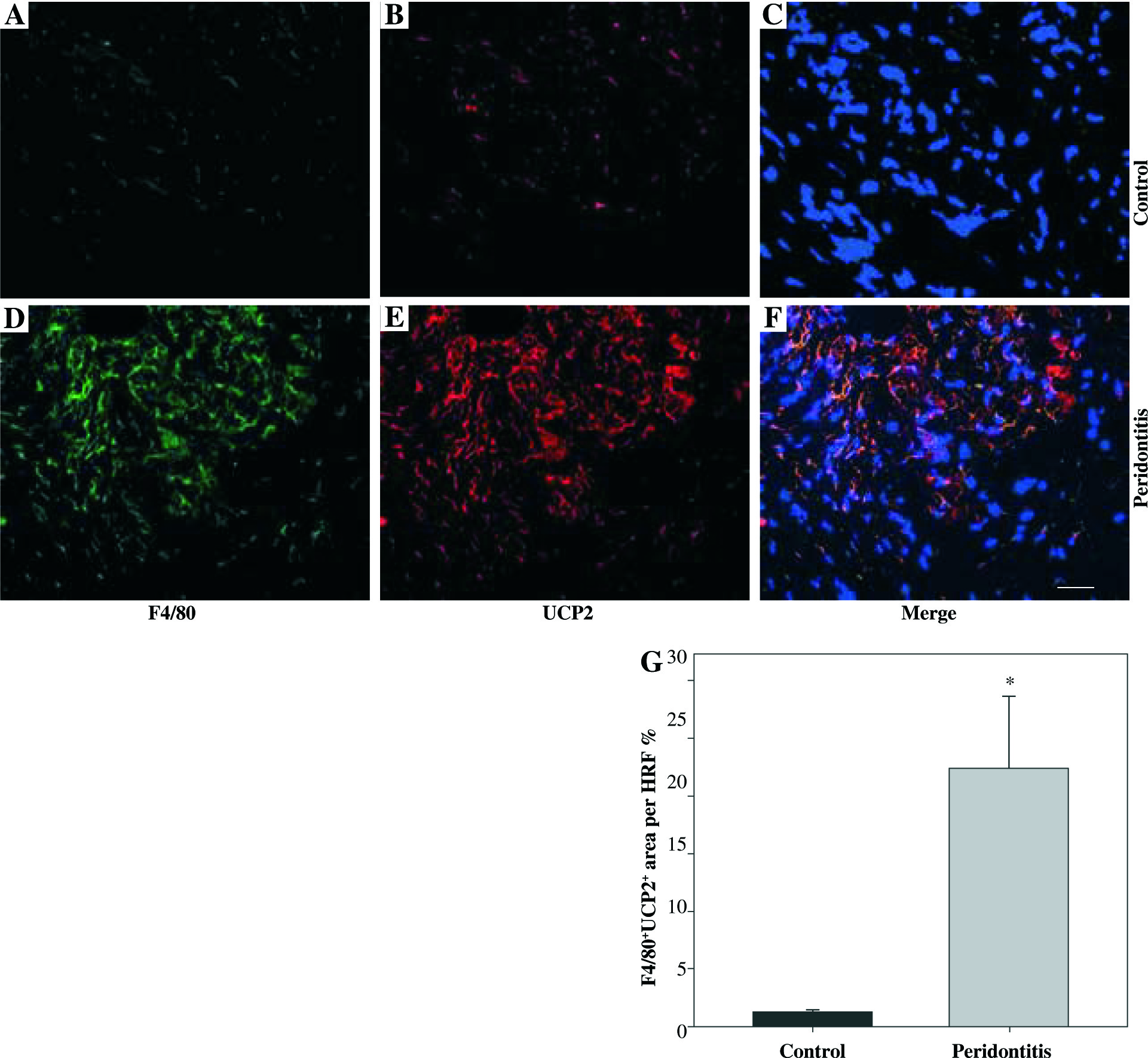
We observed macrophage infiltration in human periodontal tissues with periodontitis. Next, we collected tissue biopsies from patients undergoing surgery, and used immunofluorescence staining for F4/80 (a murine macrophage-restricted cell surface glycoprotein) and iNOS to identify M1 macrophages in gingival tissues. The H&E staining indicated that infiltrating inflammatory cells were increased in human periodontal tissues affected with periodontitis (Figs. 1A and 1B). In addition, F4/80 and iNOS double staining showed that M1 macrophages were increased in the gingival tissues with periodontitis (Figs. 1C-1I).Further immunofluorescence staining with F4/80 and UCP2 showed that infiltrating macrophages in the gingiva with periodontitis were UCP2-positive (Figs. 2A-2E). The UCP2-positive macrophages were increased in the periodontal tissues (Fig. 2G).
Fig. 1
Increased M1 macrophages infiltrating human gingival tissues with periodontitis. (A, B) H&E staining of human gingival tissue: (A) normal control gingiva, (B) gingival tissue with periodontitis. (C-H) Representative images showing the induction of pro-inflammatory macrophages in gingiva with periodontitis: (C-E) normal control gingiva, and (F-H) gingival tissue with periodontitis. (C, F) Immunofluorescent staining for F4/80, (D, G) immunofluorescent staining for iNOS, (E, H) cell nucleus (scale bars 100 μm). (I) The F4/80+ iNOS+ area in the high power field (HPF) (n = 10, * p < 0.05)
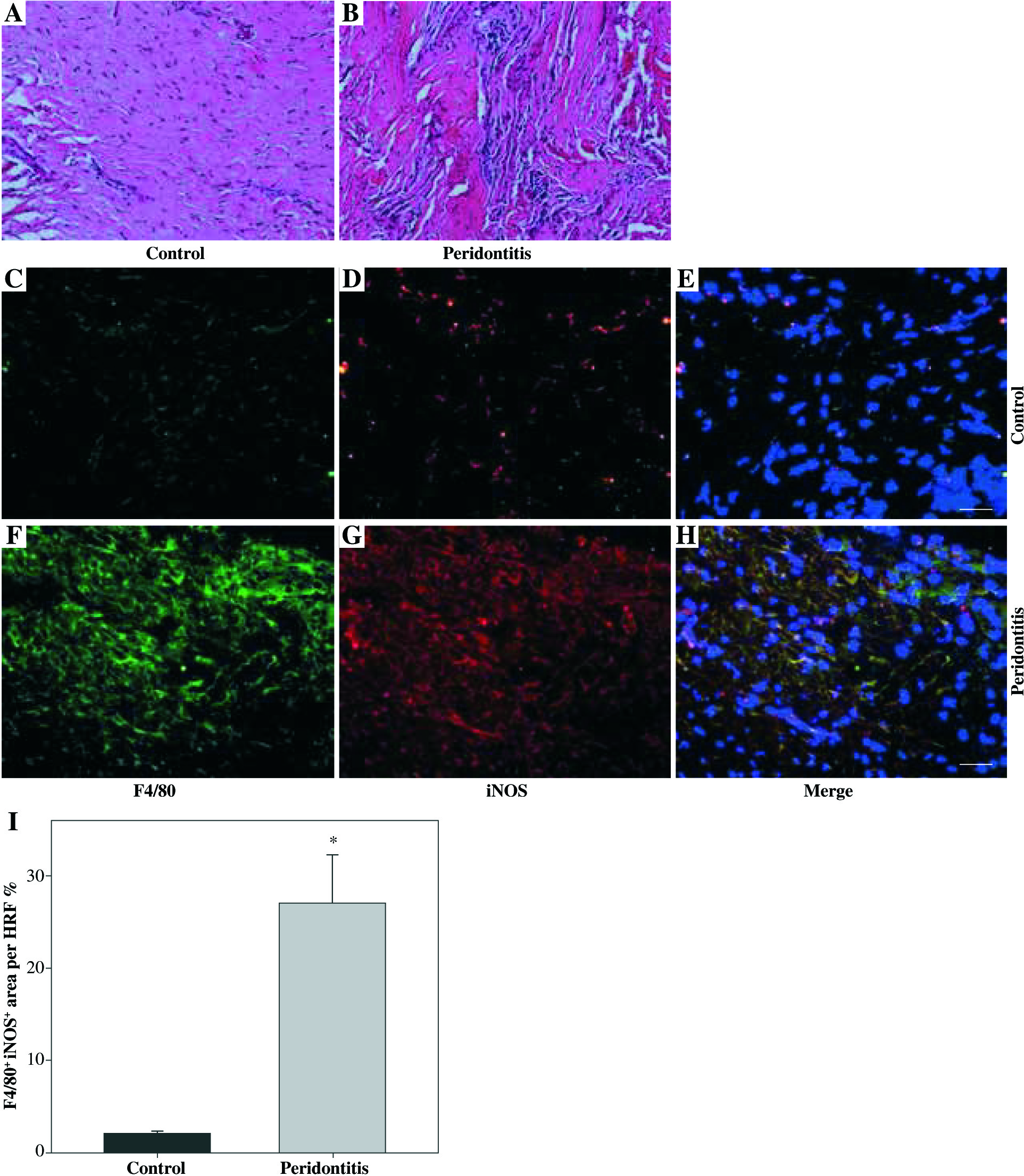
Fig. 2
UCP2 is upregulated in the macrophages in human periodontal tissues with periodontitis. (A-F) Representative micrographs showing immunofluorescent staining for F4/80 and UCP2 in human periodontal tissues: (A-C) normal control gingiva, (D-F) gingival tissue with periodontitis. (A, D) Immunofluorescent staining for F4/80, (B, E) immunofluorescent staining for UCP2 (scale bars 100 μm). (G) The F4/80+UCP2+ area in the high power field (HPF) (n = 10, * p < 0.05)
Macrophage UCP2 deficiency aggravates Pg-LPS-induced periodontitis
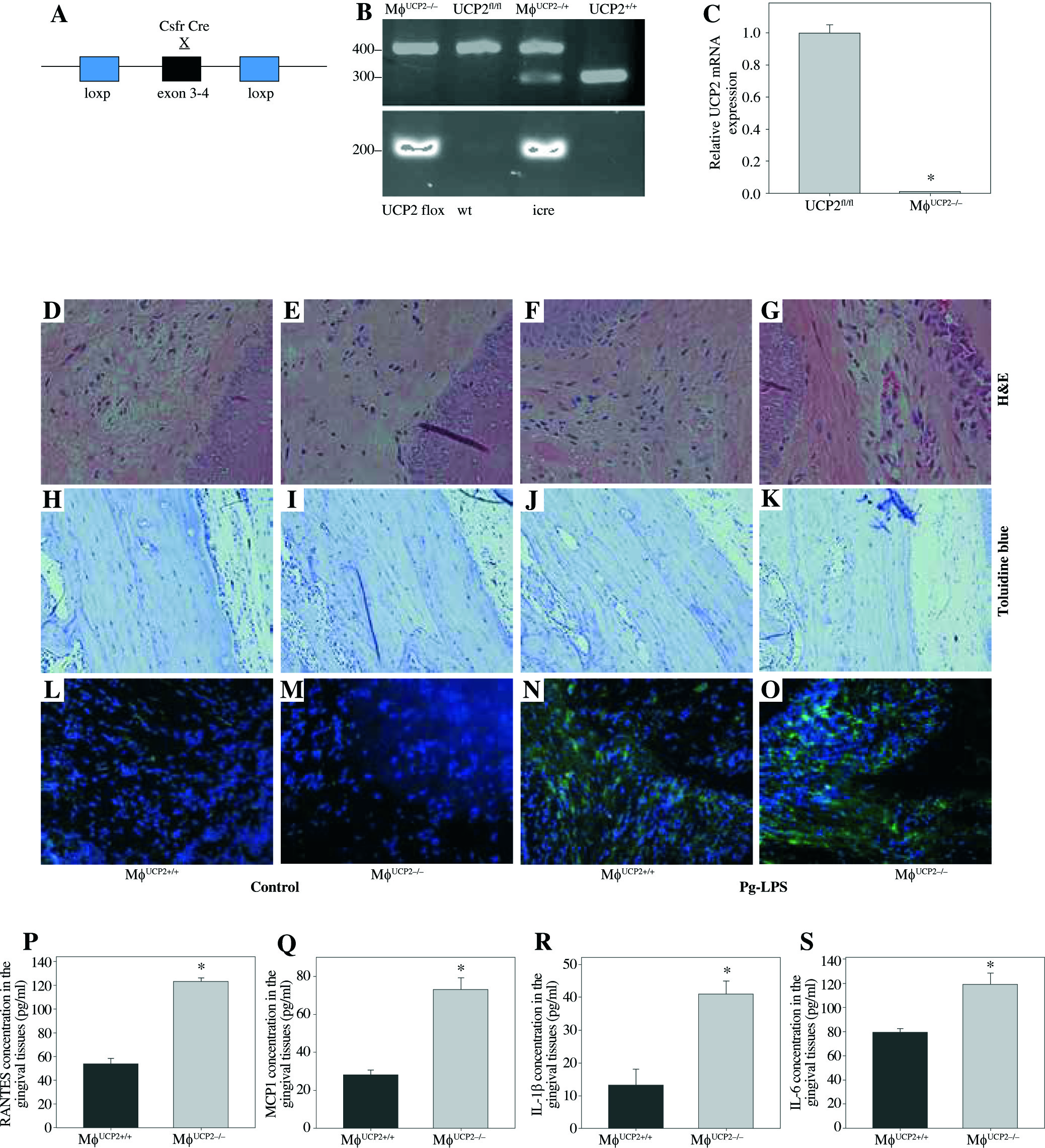
To examine the contribution of UCP2 to periodontitis, we created conditional knockout mice, in which the UCP2 gene was specifically deleted in macrophages by using the Cre-LoxP system (Figs. 3A and 3B). Mice with macrophage deletion of UCP2 were generated by intraperitoneal injection of tamoxifen for 5 consecutive days in UCP2 knockout mice (MφUCP2–/–) and their littermates. Real-time PCR analysis showed that the UCP2 expression was decreased in the peritoneal macrophages from MφUCP2–/– mice (Fig. 3C). Two days after the last tamoxifen injection, mice were injected with Pg-LPS in the mesial gingiva every two days. H&E staining showed that Pg-LPS injection could increase inflammatory cells infiltrating gingiva of MφUCP2–/– mice compared to MφUCP2+/+ mice (Figs. 3D-3G). Toluidine blue staining further indicated that oral bone was significantly disorganized, and the structure and continuity lost in Pg-LPS treated MφUCP2–/– mice compared with MφUCP2+/+ mice (Figs. 3H-K). Macrophage infiltration was also increased in gingival tissues of MφUCP2–/– mice injected with Pg-LPS (Figs. 3L-3Q). We also analyzed the cytokine levels in the gingiva of Pg-LPS-injected mice. The expression of RANTES and MCP-1 in the gingiva of MφUCP2–/– mice were twice more than that of MφUCP2+/+ mice (Figs. 3P-3Q). The production of IL-1β and IL-6 was also increased in the gingiva of MφUCP2–/– mice compared with wild type mice (Figs. 3R-3S). These data suggest that ablation of UCP2 in macrophages increases M1 macrophage infiltration and exacerbates periodontitis.
Fig. 3
Ablation of UCP2 in macrophages exacerbates mice periodontitis induced by Pg-LPS. (A) Breeding strategy for generating macrophage-specific UCP2 knockout mice (MφUCP2–/–mice). (B) Genotyping of the mice by PCR analysis of genomic DNA. (C) Real-time PCR analysis of UCP2 expression in peritoneal macrophage in MφUCP2–/– mice and control littermates. (D-K) Representative images for H&E and toluidine blue staining in mouse periodontal tissues. (L-O) Representative micrographs showing immunofluorescent staining for F4/80 in mice periodontal tissues. (P-S) The concentration of RANTES, MCP-1, IL-1β, and IL-6 in gingiva was measured using Milliplex Map assays (n = 5, * p < 0.05)
Macrophage-specific UCP2 knockout promotes pro-inflammatory M1 macrophage transformation under Pg-LPS treatment
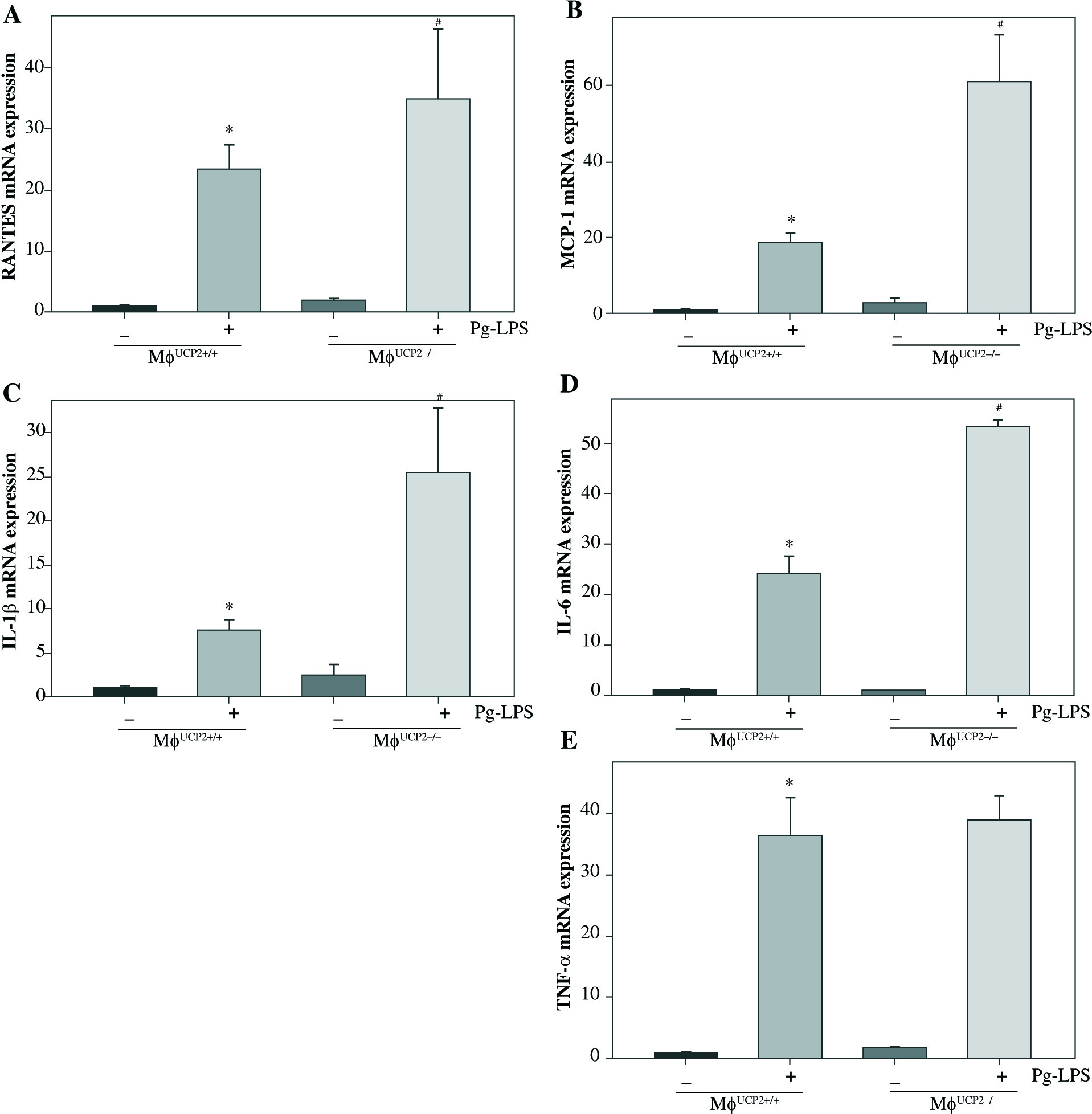
To further determine the involvement of UCP2 in macrophage activation in response to Pg-LPS, we used primary bone marrow-derived macrophage (BMM) cultures. BMMs from MφUCP2–/– mice and Mφ–UCP2+/+ mice were stimulated with Pg-LPS for 24 hours. The expression of CD86 was analyzed by flow cytometry (Fig. 4). The population of F4/80+CD86+ was increased in the Pg-LPS-treated UCP2–/– macrophages. Next, we detected MCP-1, RANTES, IL-1β, IL-6, and TNF-α mRNA by real-time PCR. The mRNA levels of the chemokines MCP-1 and RANTES in UCP2–/– macrophages were higher than UCP2+/+ macrophages after Pg-LPS treatment (Figs. 5A and 5B). IL-1β and IL-6 mRNA levels were significantly increased in UCP2–/– macrophages compared with UCP2+/+ macrophages stimulated by Pg-LPS (Figs. 5C and 5D). However, the TNF-α level in UCP2–/– macrophages was comparable with that in UCP2+/+ macrophages (Fig. 5E).
Fig. 4
Ablation of UCP2 in macrophages amplifies Pg-LPS-induced M1 macrophage polarization. FACS analysis showing M1 macrophage polarization (F4/80+ CD86+). (A) UCP2+/+ control, (B) UCP2+/+ Pg-LPS, (C) UCP2–/– control, and (D) UCP2–/– Pg-LPS
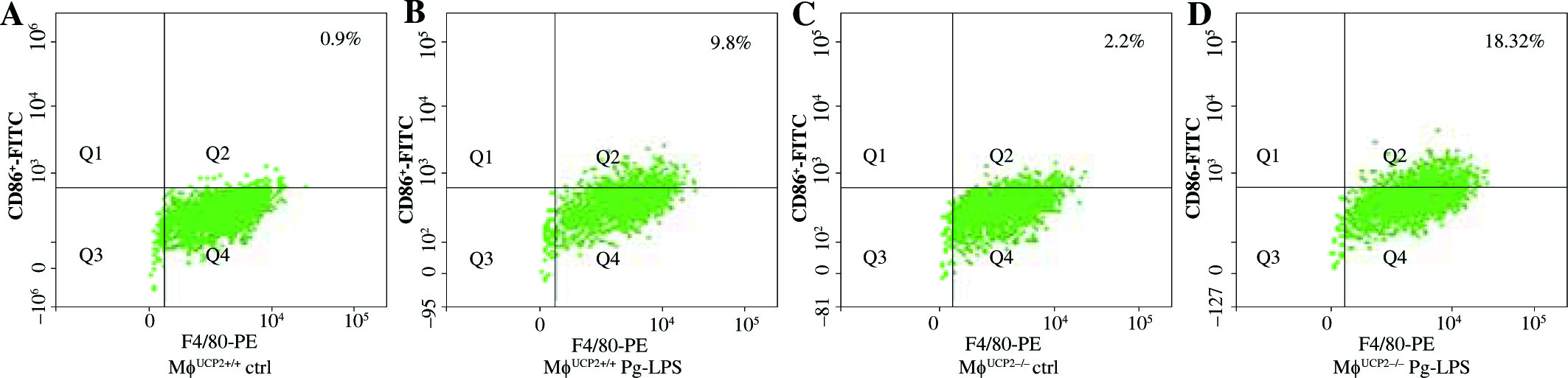
Fig. 5
Deletion of UCP2 expression in BMMs aggravates macrophage M1 polarization induced by Pg-LPS. BMMs isolated from MφUCP2–/– mice and control littermates were treated with Pg-LPS for 24 hours. Real-time PCR analysis showing the mRNA levels of (A) RANTES, (B) MCP-1, (C) IL-1β, (D) IL-6, and (E) TNF-α, * p < 0.05 vs. control, # p < 0.05 vs. UCP2+/+ macrophages after Pg-LPS treatment (n = 3)
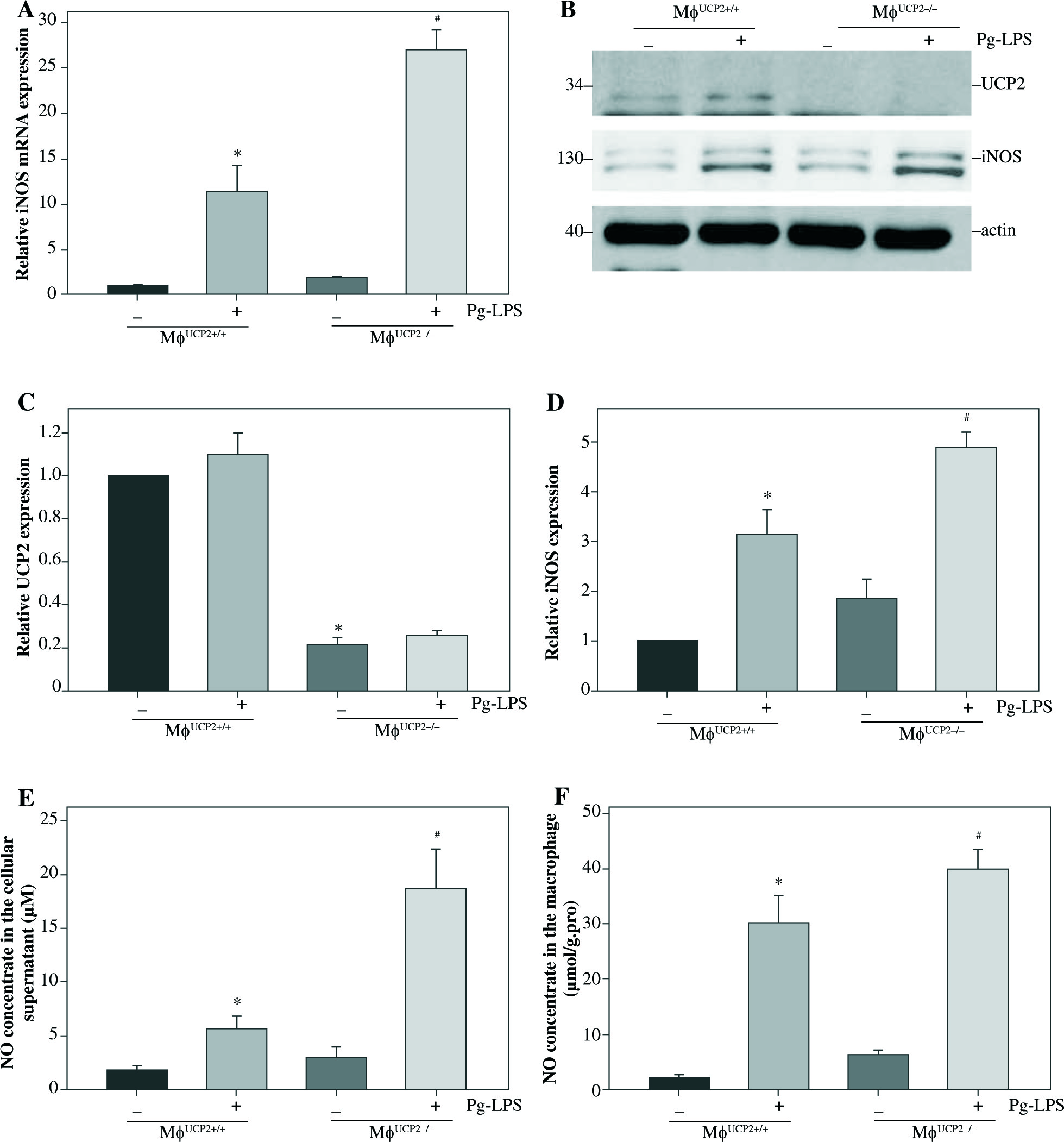
Then, we analyzed the expression of iNOS between UCP2–/– macrophages and UCP2+/+ macrophages. The mRNA and protein levels of iNOS were both highly expressed in UCP2–/– macrophages under Pg-LPS stimulation (Figs. 6A-6D). The NO levels in the macrophage and supernatant were increased in Pg-LPS-treated UCP2–/– macrophage compared with Pg-LPS-treated UCP2+/+ macrophage (Figs. 6E and 6F). These data suggest that UCP2 plays a critical role in Pg-LPS-induced M1 macrophage polarization.
Fig. 6
iNOS expression is elevated in UCP2 knockout BMMs upon stimulation with Pg-LPS. BMMs isolated from MφUCP2–/– mice and control littermates were treated with Pg-LPS for 24 hours. (A) Real-time PCR analysis showing mRNA levels of iNOS, * p < 0.05 vs. control, # p < 0.05 vs. UCP2+/+ macrophages after Pg-LPS treatment (n = 3). (B) Protein levels of UCP2 and iNOS expression detected by Western blotting. (C) Semi-quantitative analysis of UCP2 expression, * p < 0.05 vs. UCP2+/+ macrophages (n = 3). (D) Semi-quantitative analysis of iNOS expression, * p < 0.05 vs. control, # p < 0.05 vs. UCP2+/+ macrophages under Pg-LPS stimulation (n = 3). The NO levels in the (E) cell supernatant and (F) macrophages: * p < 0.05 vs. control, # p < 0.05 vs. UCP2+/+ macrophages under Pg-LPS stimulation (n = 5)
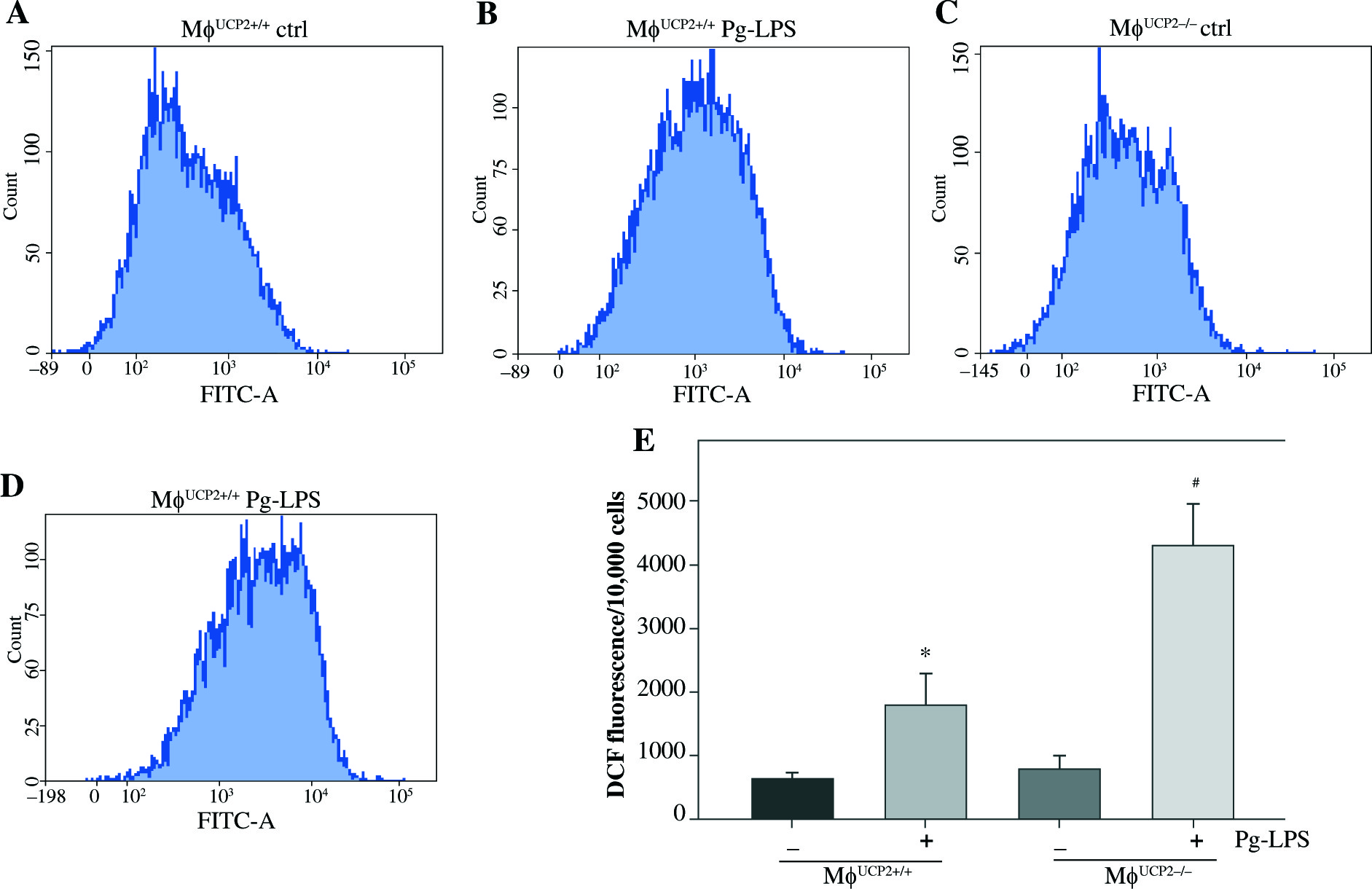
UCP2 controls Pg-LPS-induced ROS production in macrophages
UCP2 has been reported as a key regulator of ROS production in macrophages. ROS production by mitochondria was analyzed using the superoxide sensitive mitochondrial fluorescent probe DCHA-DA, and we found that Pg-LPS could increase ROS production, while knockout of UCP2 further enhance ROS production in macrophages (Fig. 7).
Fig. 7
Effects of UCP2 knockout on intracellular ROS production in BMMs stimulated by Pg-LPS. BMMs isolated from MφUCP2–/– mice and control littermates were treated with Pg-LPS for 24 hours. The level of intracellular ROS was measured with DCFH-DA staining. The DCFH-DA fluorescence abundance was detected by flow cytometry, * p < 0.05 vs. control, # p < 0.05 vs. UCP2+/+ macrophages after Pg-LPS treatment (n = 4)
UCP2 regulates the proliferation and migration of macrophages
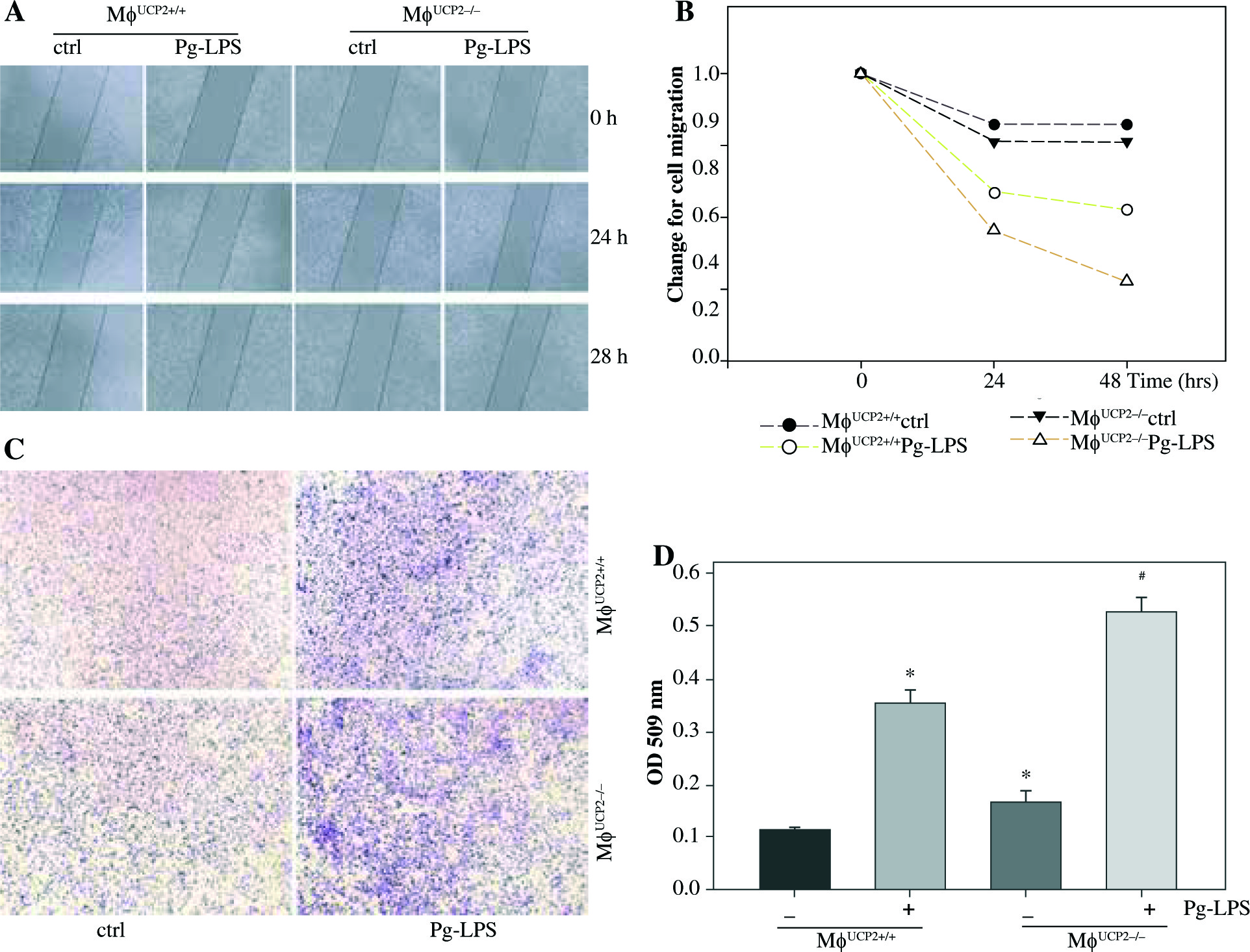
Macrophage proliferative activity was analyzed by CCK-8 assay. M-CSF induced macrophage proliferation in WT macrophages. Knockdown of UCP2 expression amplified the proliferative ability of macrophages at the same dose of M-CSF treatment (Fig. 8).
Fig. 8
Ablation of UCP2 promotes M-CSF induced BMM proliferation. BMMs isolated from MφUCP2–/– mice and control littermates were incubated with M-CSF (10 ng/ml) for 48 hours. Cell proliferation rates were measured by CCK-8 assay, * p < 0.05 vs. control, # p < 0.05 vs. UCP2+/+ macrophages incubated with M-CSF (n = 6)

We used an in vitro monolayer scratch assay and the Boyden chamber transwell assay to detect macrophage migration. The wound healing test showed that the migration ability of UCP2–/– BMMs was higher than UCP2+/+ BMMs after Pg-LPS stimulation (Figs. 9A and 9B). Then, we checked if specific knockout of UCP2 in macrophages could exacerbate migration of macrophages under Pg-LPS treatment. The Boyden chamber migration assay of cultured BMMs also exhibited that the number of UCP2–/– BMMs that migrated through the porous membrane was higher than UCP2+/+ BMMs following Pg-LPS treatment (Figs. 9C and 9D). Thus, these data indicate that specific knockout of UCP2 in macrophages displays higher proliferation and migration ability than wild type macrophages.
Fig. 9
UCP2 mediates macrophage migration. Primary macrophages from bone marrow were treated with Pg-LPS for various times as indicated. (A) Representative images for wound healing test. (B) Quantitative analysis of macrophage motility from wound healing test. (C) Representative images of the Boyden chamber migration assay in BMMs. (D) Graph showing the relative abundance of crystal violet stained in the Boyden chamber. * p < 0.05 vs. control, # p < 0.05 vs. UCP2+/+ macrophages under Pg-LPS stimulation (n = 3)
Discussion
In this study, we found that the expression of UCP2 was upregulated in the infiltrating macrophages in gingival tissues with periodontitis. In addition, macrophage-specific knockout of UCP2 could exacerbate inflammatory responses in gingiva infected with Pg-LPS. The loss of UCP2 enhanced the abilities of proliferation, migration, and pro-inflammatory cytokine excretion in macrophages.
Macrophages have been intensively studied in recent years, and divided into two categories. M1 macrophages (classically activated macrophages) express pro-inflammatory cytokines including TNF-α, IL-1β, IL-6, and high levels of ROS, whereas M2 macrophages express anti-inflammatory cytokines and growth factors such as IL-10 and TGF-β [22]. Macrophages play a significant role in periodontal inflammation and bone resorption and degradation [23, 24]. In this study, we assayed the expression of macrophages in tissues with periodontitis. We found that M1 macrophages were evidently increased in periodontitis-affected oral mucosa (Fig. 1). Previous studies in adult or aged periodontitis patients found that M1 macrophage-related gene expression was more significant than that of M2 macrophages [25]. In the Porphyromonas gingivalis-induced periodontitis mice model, the pro-inflammatory M1 macrophages were infiltrating in the early phase and contributed to alveolar bone resorption [26]. Ting et al. found that both M1 and M2 macrophages were enhanced in periodontal inflammation, and a switch from M2 to M1 was a critical mechanism in alveolar bone loss [27]. The activation of M1 macrophages with a high level of pro-inflammatory cytokines and ROS may contribute to the initiation and maintenance of periodontitis.
UCP2 is a mitochondrial anion carrier protein, which is located in the mitochondria inner membrane. UCP2 is wildly expressed in immune cells including lymphocytes and phagocytes, and plays a critical role in the immune response to various diseases like diabetes, cancer, and infectious diseases [17, 28, 29]. In this study, we created macrophage-specific UCP2 knockout mice (MφUCP2–/– mice) by crossing UCP2fl/fl mice with csf1r-iCre mice. As expected, tamoxifen injection for 5 days decreased the expression of UCP2 in isolated macrophages (Fig. 3C). Ablation of UCP2 expression in macrophages had no effect on survival rate and body weight in mice. In addition, there was no obvious change in the gingiva histology between MφUCP2–/– mice and their control littermates. When these mice were challenged with Pg-LPS, we found that macrophage infiltration, cytokines secretion, and bone destruction were more sensitive in MφUCP2–/– mice. UCP2 has been found increased in several injury such as acute kidney injury, diabetic nephropathy, and myocardial infarction [30-32]. Increased expression of UCP2 can protect cells against injury as an adaptive response toward lowering the ROS production [32-35]. In this study, we found increased UCP2 expression in the inflamed gingival tissue with periodontitis. Combined with the results that knockout UCP2 expression in macrophage aggravates the injury of periodontitis, we believe that the increased UCP2 might be a compensation against injury during periodontitis.
For years, it has been accepted that UCP2 is the main regulator of ROS production; it dissipates the proton gradient generated across the mitochondrial inner membrane, thus reducing mitochondrial superoxide production [36, 37]. UCP2 expression in differentiated macrophages was involved in ROS production [38]. Macrophages with UCP2 deficiency promotes the release of pro-inflammatory cytokines and ROS production under LPS stimulation [15, 16, 28]. Many published works on periodontitis have emphasized the essential role of ROS in tissue destruction [39, 40]. For instance, serum ROS levels were positively correlated with immunoglobulin G antibodies to periodontal pathogens [41]. ROS also exert direct effects on bone destruction, like induction of apoptosis of gingival fibroblasts, degradation of matrix [42], and promotion of alveolar bone degeneration through their role in osteoclastogenesis [43]. Here, we also observed increased ROS levels and expression of cytokines like IL-1β and IL-6 in UCP2 knockout macrophages under Pg-LPS treatment. Our data is consistent with previous observations of heightened inflammation response in UCP2 knockout macrophages. The role of regulation of ROS production by UCP2 could be a critical factor that contributes to Pg-LPS-induced periodontitis.
The expression of iNOS (inducible nitric oxide synthase) was also upregulated in Pg-LPS stimulated UCP2-ablated macrophages (Fig. 6). iNOS is an enzyme that catalyzes the production of nitric oxide (NO), which is regarded as harmful, and involved in the pathogenesis of inflammatory and autoimmune diseases. NO production by iNOS in macrophages plays a critical role in various diseases [44]. Bai et al. proved that the generation of NO was elevated in UCP2 knockout macrophages in response to LPS challenge, and contributed to persistent NK-κB activation [15]. UCP2 overexpression in macrophages markedly reduced NO production by decreasing iNOS expression [14]. It has been accepted that iNOS levels are elevated in gingiva with periodontitis [45]. In addition, increasing iNOS levels were correlated with severe inflammation in periodontal tissues [46]. Therefore, it is possible that greater ROS and NO production in UCP2 knockdown macrophages may result in more severe inflammatory response related to Pg-LPS-induced periodontitis.
Early studies using UCP2 knockout mice revealed that the ablation of the UCP2 gene could enhance the proliferation and migration ability of lymphocytes [32]. Additionally, enhanced recruitment of macrophages was found in UCP2 knockout mice [16, 17]. We found that the expression of chemokines MCP-1 and RANTES as well as the migration rate was enhanced in Ucp2-/- macrophages under Pg-LPS treatment. This indicates better ability of Ucp2-/- macrophages to recruit immune cells and migrate to gingiva with periodontitis.
Recent studies have highlighted that macrophage proliferation and activation were dependent on metabolic reprogramming [27, 30, 35]. Glycolysis was thought to be the metabolic basis of pro-inflammatory macrophage activation. UCP2 serving as a mitochondrial carrier protein was reported to alter the yield of ATP synthesis, thus exerting a central role in cellular metabolism [33, 34, 47]. UCP2 was reported to decrease mitochondrial respiration and enhance aerobic glycolysis in numerous pathophysiological conditions [48-50]. In contrast, there were opposite results supporting that glycolysis was enhanced in UCP2 knockout cells [51-53]. Thus, whether regulation of metabolism by UCP2 is involved in periodontitis-related macrophage activation needs further investigation.