
Introduction
Chronic lymphocytic leukaemia (CLL) is one of the most common types of lymphoid proliferation, with a median age at diagnosis of 70 years. In 2023, it is estimated that there will be 18,740 new cases of CLL, and an estimated 4,490 people will die of this disease. It represents 1.0% of all new cancer cases in the U.S. [1]. It usually occurs in elderly patients and has a highly variable clinical course [2]. Chronic lymphocytic leukaemia is a mainly due to the accumulation of neoplastic clusters of differentiation (CD) 5+ B-lymphocytes with a typical CD19+ and CD23+ immunophenotype [3–9]. In CLL, lactate dehydrogenase (LDH) and β2-microglobulin levels are usually within normal limits and tumour cells are Zeta-chain-associated protein kinase 70-negative (ZAP-70) [10].
Chronic lymphocytic leukaemia can transform to a more aggressive form, most commonly diffuse large B-cell lymphoma (DLBCL), known as Richter transformation (RT) [11–13]. Leukaemic transformation is initiated by specific genomic alterations that disrupt the regulation of proliferation and apoptosis in clonal B-cells [2]. Several parameters particularly higher disease stage, shorter timing of lymphocyte doubling, unmutated immunoglobulin heavy chain status and higher expression of ZAP-70 [14], deletion of chromosome 17p or 11q, activation of oncogenes such as NOTCH-1 and c-MYCN, inactivation of onco-suppressors such as TP53 and CDKN2A [15], high expression of CD38 in lymph-nodes [15–17], are associated with a worse prognosis of the disease course. Richter transformation is characterized by the confluent growth of large cells. Patients who develop RT have a poor prognosis, with a median overall survival (OS) of < 12 months despite intensive chemoimmunotherapy [14].
An accelerated phase of CLL (aCLL) can be observed, which correlates with subsequent transformation to DLBCL and may represent an early stage of transformation [11]. Accelerated CLL is a rare disease entity as it represents less than 1% of all reported cases of CLL. In aCLL, we can sometimes observe similar clinical signs, functional status, and clinical stage as in patients with CLL, however, patients with aCLL often have higher serum LDH levels [18] and elevated β2-microglobulin levels [19], and their tumour cells are ZAP70-positive. Moreover, it is most likely an underdiagnosed entity due to its rarity and the non-standardized practice of lymph node biopsy in CLL 19, and a biopsy is key to making aCLL diagnosis. The reason for tissue biopsy in patients with CLL is usually the suspicion of transformation to aggressive lymphoma (e.g., general symptoms, rapid lymph node enlargement, extensive extranodal involvement, or increased serum LDH levels) [4]. The early detection of aCLL is important as it can result in change in disease management [19]. New diagnostic methods are being explored to identify CLL at an earlier stage, alongside new treatment strategies, to maximize treatment effectiveness for patients during this phase of the illness. The aim of this review is to present the diagnostic criteria, innovative diagnostics for aCLL.
Material and methods
This narrative review aims to raise awareness of the existence of aCLL among clinicians, as it is an underdiagnosed entity due to its rarity and the non-standardized practice of lymph node biopsy in CLL with subdivision of the data into three main categories:
phases of CLL,
aCLL,
diagnostic criteria, innovative diagnostics, future clinical perspectives for aCLL.
For this purpose, we conducted an extensive literature search, querying PubMed, the most used database worldwide, from 2000 to May 2025, gathering the large part of indexed journals. All the queries and the research items were defined and developed by four experienced authors, dividing the tasks according to the authors’ expertise. Literature data analysis was based on literature indexed in the PubMed and Scopus databases. The initial criterion for inclusion of papers was the analysis of abstracts; if the paper was relevant to the aim of the study, further analysis was undertaken; preference was given to recent papers, especially those published between 2018–2025. The data retrieval process is illustrated in Figure 1.
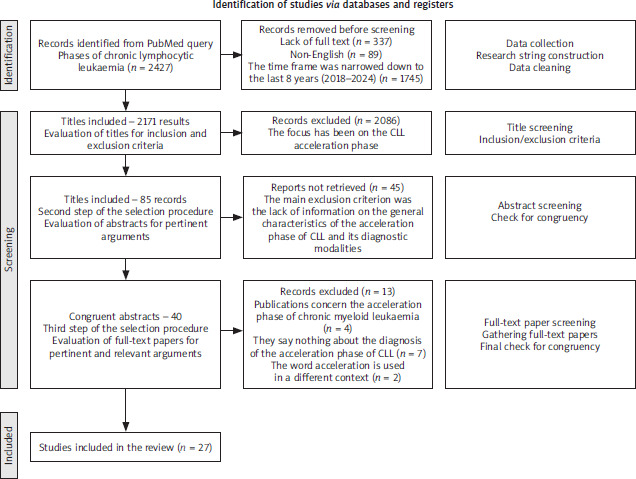
Acceleration of chronic lymphocytic leukaemia
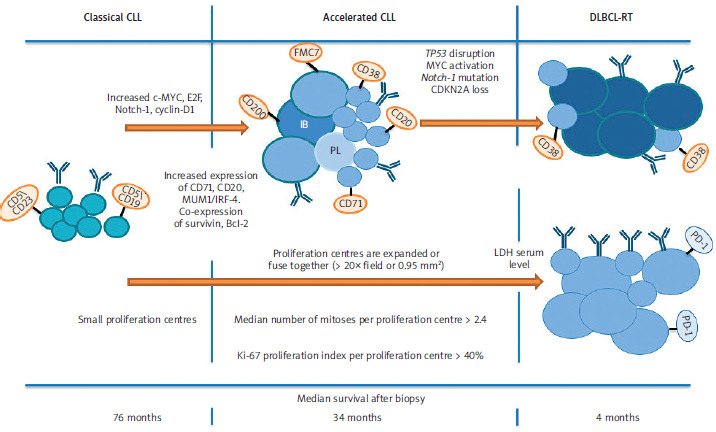
In aCLL, we can sometimes observe similar clinical signs, functional status, and clinical stage as in patients with CLL. However, patients with aCLL often have higher serum LDH levels [18] and elevated β2-microglobulin levels [19], and their tumour cells are ZAP70-positive. Patients with aCLL had significantly higher frequencies of anaemia and thrombocytopenia, splenomegaly and more advanced Rai stages [20]. In contrast, patients with unequivocal disease transformation (RT) are more symptomatic and have a lower performance status, higher serum LDH levels, and higher uptake of positron emission tomography/computed tomography (PET/CT) [18]. Several prognostic factors such as non- mutated IGHV loci, high ZAP70 and CD38 expression indicate an unfavourable prognosis [21]. According to Gine et al. [4] study, aCLL showed a poor prognosis (with a median survival after biopsy of 34 months) compared with CLL (a median survival after biopsy of 76 months) with statistical significance while the difference between the prognosis of aCLL and RT (with a median survival after biopsy of 4.3 months) was not significant (Figure 2). This shows that the prognosis of aCLL is intermediate between CLL and RT [22, 23]. Moreover, the poor prognosis of this entity was suggested by a predominance of poor prognostic markers including an increasing number of p53-positive cases in aCLL compared to standard CLL [19, 24]. In a study by Gine et al. [4] fluorescence in situ hybridization (FISH) revealed no difference in the distribution of unfavourable cytogenetic abnormalities between the three groups (typical CLL, aCLL, and RT).
Figure 2
Major histological differences between classical chronic lymphocytic leukaemia, accelerated chronic lymphocytic leukaemia, and Richter transformation
Bcl-2 – B-cell lymphoma 2, CD5 – cluster of differentiation 5, CD19 – cluster of differentiation 19, CD20 – cluster of differentiation 20, CD23 – cluster of differentiation 23, CD38 – cluster of differentiation 38, CD71 – cluster of differentiation 71, CD200 – cluster of differentiation 200, CDKN2A – cyclin-dependent kinase inhibitor, c-MYC – myelocytomatosis oncogene, CLL – chronic lymphocytic leukaemia, cyclin-D1 – D-type cell-cycle regulator of G1 phase, DLBCL-RT – diffuse large B-cell lymphoma-Richter transformation, E2F – family of transcription factor, FMC7 – conformational epitope on the CD20 molecule, IB – B lymphocyte, IRF4 – interferon regulatory factor 4, Ki-67 – marker of proliferation Kiel 67, LDH – lactate dehydrogenase, MUM1 – multiple myeloma oncogene-1, Notch-1 – neurogenic locus notch homolog protein 1, PD-1 – programmed cell death protein 1, PL – prolymphocytes, TP53 – transformation-related protein 53 Patients with classical chronic lymphocytic leukaemia have small proliferation centres and low proliferative index. In accelerated chronic lymphocytic leukaemia, following activation of c-MYC, E2F, Notch-1, cyclin-D1 and increased expression of CD71, CD20, CD38, CD71, CD200, FMC7, MUM1-IRF-4.
Peripheral blood smear
Liu et al. [25] suggest that peripheral blood smear review for increased prolymphocytes and/or atypical lymphocytes in patients with CLL may help identify patients with a higher pretest probability of transformation-related protein (TP53) abnormalities, which may be helpful in resource-limited settings. Atypical lymphocytes were defined as > 10% of the lymphocytes with marked nuclear irregularities (including cleaved nuclei, binucleate cells, and cells with micronuclei) or lymphocytes that were large in size with less condensed chromatin, small or absent nucleoli, and moderate-to-abundant cytoplasm [25]. Regarding periphe- ral blood morphology in CLL patients with TP53 alterations, 40% demonstrated atypical lymphocytes, accounting for > 10% of the lymphocytes, 71% had atypical lymphocytes with marked nuclear irregularities (including cleaved nuclei, binucleate cells, and cells with micronuclei). In the remaining 29%, the atypical lymphocytes displayed a large size, less condensed nuclear chromatin, small or absent nucleoli, and moderate-to-abundant cytoplasm. However, the presence of circulating atypical lymphocytes was not prognostically relevant compared to other patients with TP53-mutated CLL [25]. There was a significant correlation between bone marrow iron storage and the percentage of conformational epitope on the CD20 molecule (FMC7) marker expression and the percentage of atypical lymphocytes in the peripheral blood [26].
Histological diagnosis
Diagnosis of accelerated CLL is based on histological evolution and was first reported by Gine et al. [4]. The reason for tissue biopsy in patients with CLL is usually the suspicion of transformation to aggressive lymphoma (e.g., rapid lymph node enlargement, extensive extranodal involvement, progressive thrombocytopenia and anaemia, autoimmune steroid-resistant cytopenias) [4]. Patients with CLL or small lymphocytic leukaemia (SLL) with clinical suspicion of disease progression, especially those with bulky disease and PET/CT maximum standardized uptake value (SUV) ≥ 5, should undergo biopsy at the site of highest metabolic uptake to establish a definitive pathological diagnosis [27]. Distinguishing CLL from aCLL or RT can be challenging, particularly in small needle-biopsy specimens. However, there are currently no standard, commonly accepted criteria for the morphological differentiation between aCLL and classic CLL [28–30]. The most common method of aCLL diagnosis is biopsy followed by histological staining. Tissue samples were analysed for the presence and size of proliferation centres (PCs), and for proliferation status as assessed by the mitotic index and marker of proliferation Kiel 67 (Ki-67) immunostaining. Enlarged nodes from patients with CLL show effacement of the lymphoid architecture with a pseudofollicular pattern of pale areas on a dark background of small cells. These pale areas correspond to PCs and are predominantly composed of clusters of prolymphocytes and paraimmunoblasts that have increased mitotic activity. Proliferation centres were also identified and delineated by the fact that they did not stain with p27. Prolymphocytes are small to medium in size, showing dispersed nuclear chromatin and often with an irregular nuclear contour [31]. Paraimmunoblasts are larger cells with dispersed chromatin, a prominent central eosinophilic nucleolus, and an expanded cytoplasm [32]. The study by Gine et al. [4] defined histological criteria for aCLL, which are enlarged and confluent PCs wider than 20 times the field of view, a median number of mitoses per PCs of 0.9 (range: 0–12 mitoses) and a median Ki-67 proliferation index per PCs of 10% (range: 0–75%). The median Ki-67 in the whole tissue section was 2% (range: 0–30%). However, in the study by Liu et al. [25] the mean Ki-67 proliferation rate in the samples was 30% (range: 10–70%). Morphologic findings in the lymph nodes involved by CLL carrying p53 alterations are expanded PCs or increased numbers of medium- and/or large-sized lymphoid cells with no expansion of PCs (“paraimmunoblastic variant”). The National Comprehensive Cancer Network (NCCN) guidelines recommend that ‘CLL with enlarged PCs’ or ‘accelerated CLL’ can be diagnosed in cases where the PCs in CLL are enlarged or fused (> 20× field or 0.95 mm2) and show a Ki-67 proliferation index > 40% or > 2.4 mitosis/PC [33].
The proliferation centres may show increased expression of Ki-67, c-MYC, family of transcription factor, Notch-1, and cyclin-D1, highlighting its role in tumour proliferation [32]. Compared to the non-PC component of CLL, cells clustered in the PCs have increased expression of the proliferation- associated markers Ki-67 and CD71; co-expression of survivin and B-cell lymphoma 2 (Bcl-2); and higher expression of CD20, interferon regulatory factor 4/multiple myeloma oncogene-1 (MUM1/IRF-4) (Figure 2) [34–36]. Absence or weak expression of CD5 or CD23, are most commonly considered as aCLL [37, 38, 60]. The assessment of additional antigens in flow cytometry, especially the CD200 glycoprotein, may facilitate the process of differential diagnosis of atypical CLL from other B-cell lymphoproliferative neoplasms [37]. The current (2022) World Health Organization (WHO) classification of haematolymphoid malignancies does not provide morphological guidelines for evaluating CLL cases with clinically suspected accelerated disease [39]. This raises the possibility of false-positive and false-negative diagnoses of aCLL. Inter-observer variability will result in different treatment decisions. There is a need for standardized guidelines (possibly by future WHO classifications) for aCLL.
Cytogenetic abnormalities
Chronic lymphocytic leukaemia with expanded PCs on morphology (as seen in aCLL) was more likely to carry 17p and 11q deletions, the TP53 mutation, and a complex karyotype [40, 41]. The deletion of 17p involving the loss of the TP53 gene and/or the mutations in TP53 are identified in 4–10% of patients at diagnosis [42, 43] but can be acquired throughout the disease course. In refractory CLL, it is 42–45% [44]. The chronic lymphocytic leukaemia phase preceding RT (which needs not always go through an acceleration phase) had a higher frequency of certain genetic alterations (i.e., del17p, del15q, and add2p). More than 50% of patients with RT have a TP53 aberration in the CLL clone before transformation [45–49]. Several studies have shown that many cases of RT appear to result from CLL with non-mutated IGHV [50–52]. Likewise, during the acceleration phase of CLL, non-mutated IGHV gene loci are observed more frequently [21]. Liu et al. [25] showed that the presence of the morphologic features suggestive of the accelerated phase had no effect on OS within the CLL group with TP53 abnormalities, demonstrating that the prognosis of both CLL and aCLL patients with mutations within the TP53 gene was equally unfavourable. However, without known TP53 abnormalities, aCLL shows a significantly worse prognosis than CLL, as shown in the study by Gine et al. [4]. Transformation-related protein mutation status testing is of value in evaluating of CLL/aCLL.
Molecular basis of accelerated chronic lymphocytic leukaemia
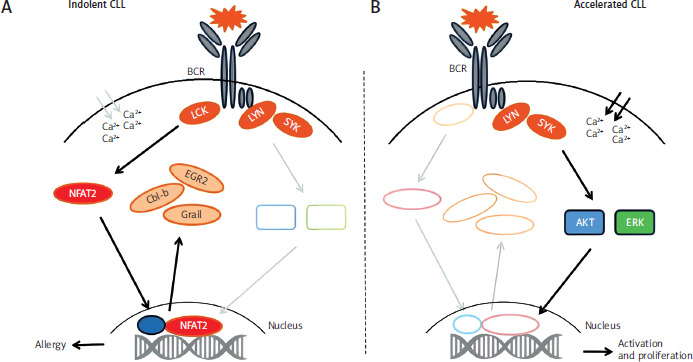
Tonic stimulation of the B-cell receptor (BCR) in indolent CLL is associated with the recruitment of lymphocyte-specific protein tyrosine kinase (LCK) and the activation of nuclear factor of activated T-cells 2 (NFAT2), which subsequently translocates to the nucleus to induce the transcription of several anergy-associated genes including Cbl-b, Grail, Egr2 as well as Lck (Figure 3, left panel). Ablation of LCK leads to loss of the anergic phenotype, loss of NFAT2 accelerates selection of non-mutant BCR, variable- diversity-joining recombination (‘BCR hyperreactivity’), BCR stimulation leads to enhanced phosphotyrosine induction and calcium flux culminating in the activation of protein kinase B and extracellular signal-regulated kinases and subsequent cell proliferation as observed in patients with aggressive forms of CLL or Richter’s syndrome (Figure 3, right panel) [53].
Figure 3
Molecular basis of accelerated chronic lymphocytic leukaemia. Putative mechanism of the role of the nuclear factor of activated T-cells 2 in the regulation of the anergic phenotype in chronic lymphocytic leukaemia
AKT – protein kinase B, BCR – B-cell receptor, Cbl-b – E3 ubiquitin-protein ligase, CLL – chronic lymphocytic leukaemia, EGR2 – early growth response protein 2, ERK – extracellular signal-regulated kinases, Grail – E3 ubiquitin ligase, Lck – proto-oncogene, LYN – tyrosine-protein kinase, NFAT2 – nuclear factor of activated T-cells 2, SYK – spleen tyrosine kinase
Trephine biopsy
It is speculated that the variable staining pattern of p53 immunohistochemistry may potentially reflect tumour heterogeneity, as well as the effect of decalcification in the bone marrow trephine biopsies and the technical artefact in the peripheral blood cell block preparation. Immunohistochemical staining with the p53 antibody has high specificity but low sensitivity. Thus, negative results should be interpreted cautiously [25]. This challenge is compounded by other variables, such as the amount of tissue present for evaluation and the need to evaluate both nodal and non-nodal sites of the disease. Artifacts of tissue processing and staining can also contribute to diagnostic uncertainty, and laboratories would be wise to mitigate this influence by optimizing protocols for lymph node specimens to ensure high quality morphologic evaluation. Accompanying this area of diagnostic uncertainty, the clinical implications of assigning these histologic categories is not always clear [54]. While genetic alterations of CLL/SLL associated with adverse clinical behaviour have been repeatedly studied, correlation of these genetic signatures with aggressive histologic features remains imperfect [41, 55]. This is compounded by molecular and cytogenetic studies of CLL/SLL, in both clinical and research settings, being commonly performed on peripheral blood or bone marrow specimens, as opposed to formalin- fixed-paraffin-embedded (FFPE) tissue [56]. Currently, there is no work on the use of trephine biopsy for the diagnosis of aCLL. However, based on our clinical experience, we believe that the use of trephine biopsy to detect CLL progression is perhaps the way of the future. As a result, we will not have to perform a lymph node biopsy procedure, which requires the involvement of other specialists and exposes the patient to various complications, reduced invasiveness, broader applicability. The procedure of trephine biopsy from the posterior superior iliac spine usually takes 10 minutes, but the procedure may take up to 30 minutes depending on other sites of biopsy, experience of the physician and co-operation of the patient [57, 58]. Despite its highly invasive and painful nature, complications are exceptionally rare and trephine biopsy is generally considered a safe and low risk procedure [59]. As correlation of genetic and histologic features remains limited, a larger study that serially examines genetic alterations in FFPE tissue in conjunction with histology, from initial diagnosis and throughout the disease course, may be helpful in establishing indicators of evolving or progressive disease to help refine clinical action points [54]. The use of trephine biopsy in the diagnosis of aCLL may be the subject of numerous studies in the coming years.
Radiological manifestations
The role of PET/CT in the diagnostic process remains unclear. Still, a higher SUV in aCLL compared to CLL has been reported. Due to the low number of described cases, it is impossible to give clear recommendations; however, considering that some cases were diagnosed during the exclusion of RT, it seems plausible that PET/CT should be performed in all cases suspected of RT, indicating the right lymph node/tissue for biopsy to establish the diagnosis. The diagnosis of aCLL/SLL does not exclude the diagnosis of RT or the possibility of its development [60].
Machine learning approaches to accelerated chronic lymphocytic leukaemia diagnosis
Machine learning is a method that has been investigated recently for its potential to diagnose aCLL. El Hussein et al. [11] designed an artificial intelligence (AI)-based tool to identify and delineate PCs based on feature analysis of the combined individual nuclear size and intensity, designated here as the heat value. It should be remembered that PCs in CLL and aCLL are dynamic environments, in which an initially minor percentage of PCs have crossed over to a more progressive state (CLL into aCLL or aCLL into RT).
Previously, authors proposed and validated an AI model based on four morphologically meaningful cellular attributes (nuclear size, intensity, cellular density and cell to nearest neighbour distance). They suggested that the value of 24 µm was the optimal nuclear size cutoff that best distinguished CLL from aCLL and RT [61]. However, with a combined analysis of nuclear size and intensity analysis, which El Hussein et al. [11] called ‘heat value’, better results were achieved. They retrospectively searched patients with haematolymphoid diseases clinically evaluated at MDACC. They randomly selected 10 CLL, 12 aCLL, and 8 RT digitised hematoxylin and eosin stained slides of excisional biopsy specimens of lymph nodes to study the mapping of PCs. Slide scanning was conducted using Aperio AT2 scanners at an optical resolution of 20× (0.50 µm/ pixel). All selected slides came from different patients, and in total we manually annotated 25, 28, and 21 regions of interest (ROI) encompassing small round PCs and confluent/expanded PCs from CLL, aCLL, and RT, respectively. The regions of interest selection was random in the three disease categories and did not target any specific areas to decrease selection bias. However, during ROI annotation, the researchers avoided areas with tissue folding, red blood cell extravasation and accumulation, and, in RT cases, areas of necrosis, as these morphologic features could affect the performance of their algorithm targeting their cells of interest [11].
Mean heat value
Using the mean heat value from the generated heat value image of all cases, CLL, aCLL, and RT can be separated with sensitive diagnostic predictive values. Areas with high heat values (in the yellow spectrum) correspond to tiles harbouring cells with increased nuclear size and mean intensity (PCs in CLL cases and expanded/confluent PCs in aCLL and RT cases). In contrast, areas with low heat values (in the blue spectrum) correspond to tiles with a decreased nuclear size and mean intensity, representing small neoplastic lymphocytes surrounding PCs. This recreation of PCs based on objective measures of nuclear attributes (size and intensity) provides a visual aid to assess the extent of large cells (with large nuclei) depicted in yellow in relation to small neoplastic lymphocytes in blue, in the three disease phases [11]. There were two optimal heat value thresholds isolated based on the F-score to achieve the best separation among the three disease phases: 0.228, below which the case was most likely to be CLL, and 0.288, above which the case was most likely to be RT. Cases with heat values ranging from 0.228 to 0.288 were most likely aCLL. There was a significant difference in the ranges of mean heat value frequencies for CLL, aCLL, and RT, which were 0.168–0.233, 0.212–0.307, and 0.261–0.353, respectively. Using this method, they were able to enhance the visualisation of the overall architectural distribution and extent of large cells in the studied ROI: confined yellow foci in PCs in CLL, confluent yellow foci representing fused PCs in aCLL, or yellow sheets replacing the vast majority of the ROIs in RT [11].
Limitations of machine learning approaches
It is important to note the limitations of this method, such as the small sample sizes and the need for external validation across multiple centres. Potential pitfalls in relation to the use of this model such as variability in slide quality, difficulties in selecting the area of interest and inter-laboratory differences in tissue processing confirm the continued lack of readiness of these tools for routine diagnostic use. In clinical practice, the distinction between CLL and aCLL in patients being evaluated for progressive disease can be challenging, particularly in core biopsy specimens, the most common sampling biopsy technique for tissue confirmation in CLL patients with features of disease progression. Evaluating such samples usually requires subspecialty expertise, and even with such expertise, distinction requires a high degree of proficiency. These challenges make CLL disease phases an ideal substrate for developing deep learning algorithms that can serve as diagnostic aids to address clinical challenges in this space [62].
A practical diagnostic pathway for patients with accelerated chronic lymphocytic leukaemia
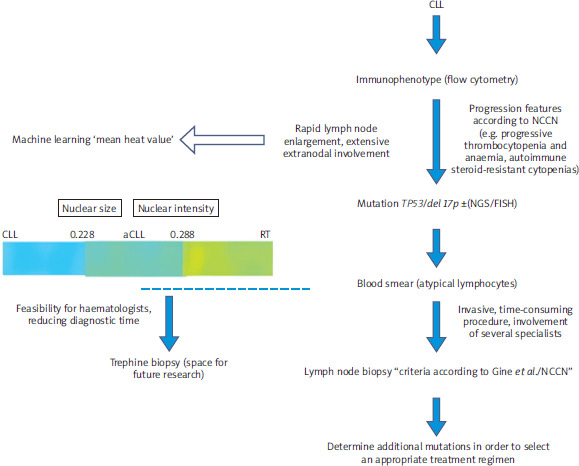
Patients diagnosed with CLL have flow cytometry performed to determine immunophenotype. Higher expression of CD71, CD20, MUM1/IRF-4, CD10, CD22, CD49d, CD81 and FMC7 and lower expression of CD5, CD23, CD148 and CD200 may indicate aCLL. It is useful to have some flow cytometry results (diagnosis of CLL, suspicion of progression) to assess changes in cell immunophenotype. When progression is suspected (rapid lymph node enlargement, extensive extranodal involvement, progressive thrombocytopenia and anaemia, autoimmune steroid-resistant cytopenias), a peripheral blood smear should be performed to show the presence of atypical lymphocytes or prolymphocytes. Their presence in patients with CLL may help identify patients with a higher pretest probability of TP53 abnormalities, which may be helpful in resource-limited settings (Figure 4). Then we should assess the presence of TP53 mutations, 17p deletion, 11p deletion, IGHV mutations and trisomy 12 by FISH or next-generation sequencing (NGS). For the diagnosis of aCLL, we should proceed with lymph node biopsy and follow the guidelines according to Gine et al./NCCN. Here we see space for trephine biopsy, which is a less invasive, less time- consuming procedure and does not require the involvement of several specialists. In a further step, we mark additional mutations, uIGHV, del15p, add2p. The increasing availability of methods based on machine learning (heat average) offers opportunities to continuously improve this model, as it may allow rapid diagnosis of patients in aCLL phase in the future, and this will enable active clinical trials dedicated to the acceleration phase. However, there are currently many limitations to the effective use of machine learning, such as the small sample sizes and the need for external validation across multiple centres.
Figure 4
Diagnostic algorithm for patients with accelerated chronic lymphocytic leukaemia and the colour distinction of the three phases [11]
aCLL – accelerated chronic lymphocytic leukaemia, CLL – chronic lymphocytic leukaemia, del 17p – deletion 17p, FISH – fluorescence in situ hybridization, NGS – next generation sequencing, NCCN – National Comprehensive Cancer Network, RT – Richter transformation, TP53 – transformation-related protein 53
Treatment of the accelerated chronic lymphocytic leukaemia
The approach to the treatment of CLL has changed considerably over the last few years. In the past, first-line treatment was chemo-immunotherapy. At present, targeted therapies in CLL utilize kinase inhibitors like those aimed at Bruton tyrosine kinase (BTK) and Bcl-2 inhibitors (venetoclax), replacing chemoimmunotherapy [63]. The presence of a non-mutated IGHV gene or del(17p), del(11q) or TP53 mutations in patients receiving chemo-immunotherapy, including fludarabine, cyclophosphamide and rituximab, is associated with poorer progression-free survival (PFS) [2, 64–67]. According to NCCN guidelines, it is participation in a clinical trial that is the recommended treatment option for aCLL, as no optimal regimen has been established to date. In the absence of a suitable clinical trial, aCLL should be treated with treatment options defined for CLL based on the presence of del(17p) or TP53 mutations.
A multicentre, retrospective study was recently conducted involving patients diagnosed with aCLL between 2013 and 2023 in thirteen Polish Adult Leukemia Study Group haematology centres. The entire cohort was divided into two groups: treatment-naïve patients, who either experienced disease acceleration at diagnosis or were observed with CLL and later accelerated during follow-up, and relapsed/refractory (patients, who exhibited acceleration after prior CLL treatment). The majority received immunochemotherapy. Twenty-two patients (20.8%) were treated with immunochemotherapy including rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) and R-CHOP-like protocols. Twenty-one (19.8%) received the bendamustine-rituximab protocol. Seventeen patients (16.0%) received fludarabine-based immunochemotherapy. Finally, twenty-three patients were treated with targeted therapy. This group included BTK inhibitors monotherapy (ibrutinib, acalabrutinib, a single patient received TL-895) and venetoclax-based regimens (obinutuzumab-venetoclax, rituximab-venetoclax, and venetoclax monotherapy). Nine patients were not treated. In a further analysis of the whole group, fludarabine-based regimens proved to be superior to bendamustine-based regimens in OS and PFS, and to R-CHOP-like protocols in both OS and PFS. There was no statistically significant difference in OS and PFS between fludarabine-based regimens and targeted therapy or in any other performed comparisons. However, the group of patients treated with treatments including both BTK inhibitors and venetoclax, either in monotherapy or in combination with rituximab or obinutuzumab was very heterogeneous. Thus, the comparison of treatment outcomes between immunochemotherapy and novel agents may be biased. It should be noted that patients in whom fludarabine-based regimens were used as first-line treatment of acceleration were statistically significantly younger than patients treated with R-CHOP-like protocols (mean age 55.15 vs. 62.4 years). There were no statistically significant differences between these groups in Eastern Cooperative Oncology Group or the number of previous lines of CLL treatment [23]. However, R-CHOP-like protocols appeared to be associated with a worse prognosis than other immunochemotherapy protocols, suggesting that their use in aCLL treatment should be limited [23, 60]. In light of the lack of specific guidelines for the diagnosis of aCLL, there are few papers on the potential treatment of aCLL. We present papers that address the treatment of aCLL, but these are mostly single clinical case reports where ibrutinib [32, 68], acalabrutinib [69], venetoclax with rituximab [70] have been used successfully (Table 1).
Table 1
Treatment of accelerated chronic lymphocytic leukaemia
| Number of patients | Method of treatment | References |
|---|---|---|
| 106, TN = 46, R/R = 60 | 22/106 R-CHOP, 21/106 B/R, 17/106 fludarabine-based immunochemotherapy, 23/106 targeted therapy (BTKi monotherapy, venetoclax-based regimens) | [23] |
| 13 | 5/13 no treatment 8/13 BTKi | [27] |
| 1 | Ibrutinib | [68] |
| 2 | Ibrutinib | [32] |
| 1 | Venetoclax, rituximab | [70] |
| 1 | Acalabrutinib | [69] |
Future perspectives
Unlike RT, clinical trials conducted so far have not included or excluded cases of aCLL/SLL within their eligibility criteria. To the authors’ knowledge, no clinical trials have yet incorporated this entity into their design; however, with growing understanding, it seems reasonable to include this disease entity in future clinical trials, as retrospective data published so far suggest it may influence treatment outcomes [60].
Conclusions
‘Accelerated’ CLL is likely to reflect the biological state of CLL tumour cells characterized by high proliferation, resulting in a more aggressive form of the disease and, consequently, a poor prognosis. The aim of this review was to raise awareness of the existence of aCLL among clinicians, as it is an underdiagnosed entity due to its rarity and the non-standardized practice of lymph node biopsy in CLL, so it is worth considering a lymph node biopsy or trephine biopsy when CLL progression is suspected. The use of trephine biopsy in the diagnosis of aCLL may be the subject of numerous studies in the coming years. Other hallmarks of aCLL should be sought based on the phenotype and genetics (e.g., TP53 mutation and deletion 17p). Using the mean heat value which consists of the size and intensity of the nucleus, CLL, aCLL, and RT can be separated with sensitive diagnostic predictive values. The development of biomedical engineering and various modern techniques based on unsupervised learning makes it easier to diagnose the disease in the acceleration phase. However, clinical trials in aCLL are needed to select an appropriate treatment regimen in these patients, especially those with TP53 mutation or deletion 17p, to achieve better aCLL treatment outcomes. Future studies might benefit from combining genomic, histologic, and radiological data to build comprehensive models of disease progression.






