
Introduction
Kleefstra-2 syndrome (OMIM #617768) is a rare neurodevelopmental disorder characterized by developmental delay, intellectual disability, autism spectrum disorders, and characteristic dysmorphic features [1]. This condition results from heterozygous pathogenic variants of the KMT2C gene, encoding a histone-lysine methyltransferase involved in epigenetic regulation of gene expression [1].
Despite recent recognition, this pathology remains exceptionally rare in the medical literature. Even in the largest international cohort reported to date (81 cases), exploration of endocrine manifestations remains insufficient, particularly concerning documentation of the somatotropic axis [2].
Growth hormone deficiency is a frequent cause of short stature in children, generally treated effectively with recombinant growth hormone administration. However, some patients present a suboptimal treatment response, raising questions about alternative diagnosis or more complex underlying pathology [3].
We present a young girl initially diagnosed with growth hormone deficiency, whose atypical clinical evolution led to the discovery of Kleefstra-2 syndrome. This observation raises important questions about potential interactions between epigenetic abnormalities and the somatotropic axis.
Case report
We report Aya E., a 12-year-10-month-old girl, referred for reevaluation of persistent short stature despite growth hormone treatment in January 2024.
Born at term to second-degree consanguineous parents, the patient presented normal birth parameters. Neurodevelopmental evolution revealed psychomotor delay with delayed walking acquisition at 21 months and delayed language development, characterized by emergence of first words around 3 years and production of simple sentences only at 5 years. These developmental disorders were accompanied by major learning difficulties requiring educational pathway adaptation.
Short stature exploration undertaken at age 8 and 6 months in October 2019 revealed significant growth retardation (height 116 cm, –2.5 SD; weight 18 kg, –2.5 SD) associated with three-year bone maturation delay (bone age 4 years for chronological age 7 years). Genetic target height calculated from parental heights was 165 cm (median). Endocrinological assessment revealed decreased serum insulin-like growth factor-1 (IGF-1) concentration at 64 ng/ml (age-specific normal range: 75–286 ng/ml) and growth hormone secretory insufficiency confirmed by pathological L-DOPA and insulin stimulation tests (GH peak < 5 ng/ml). Thyroid and adrenal explorations were normal. Pituitary magnetic resonance imaging and karyotype analysis showed no abnormalities.
At this stage of the initial evaluation, dysmorphic features were subtle, and the chief complaint was primarily short stature associated with developmental delay. This clinical presentation, combined with biochemical findings, justified the endocrinological diagnostic approach and initiation of growth hormone treatment according to standard recommendations, with recombinant somatotropin substitutive treatment initiated at 0.035 mg/kg/day at age 9 years.
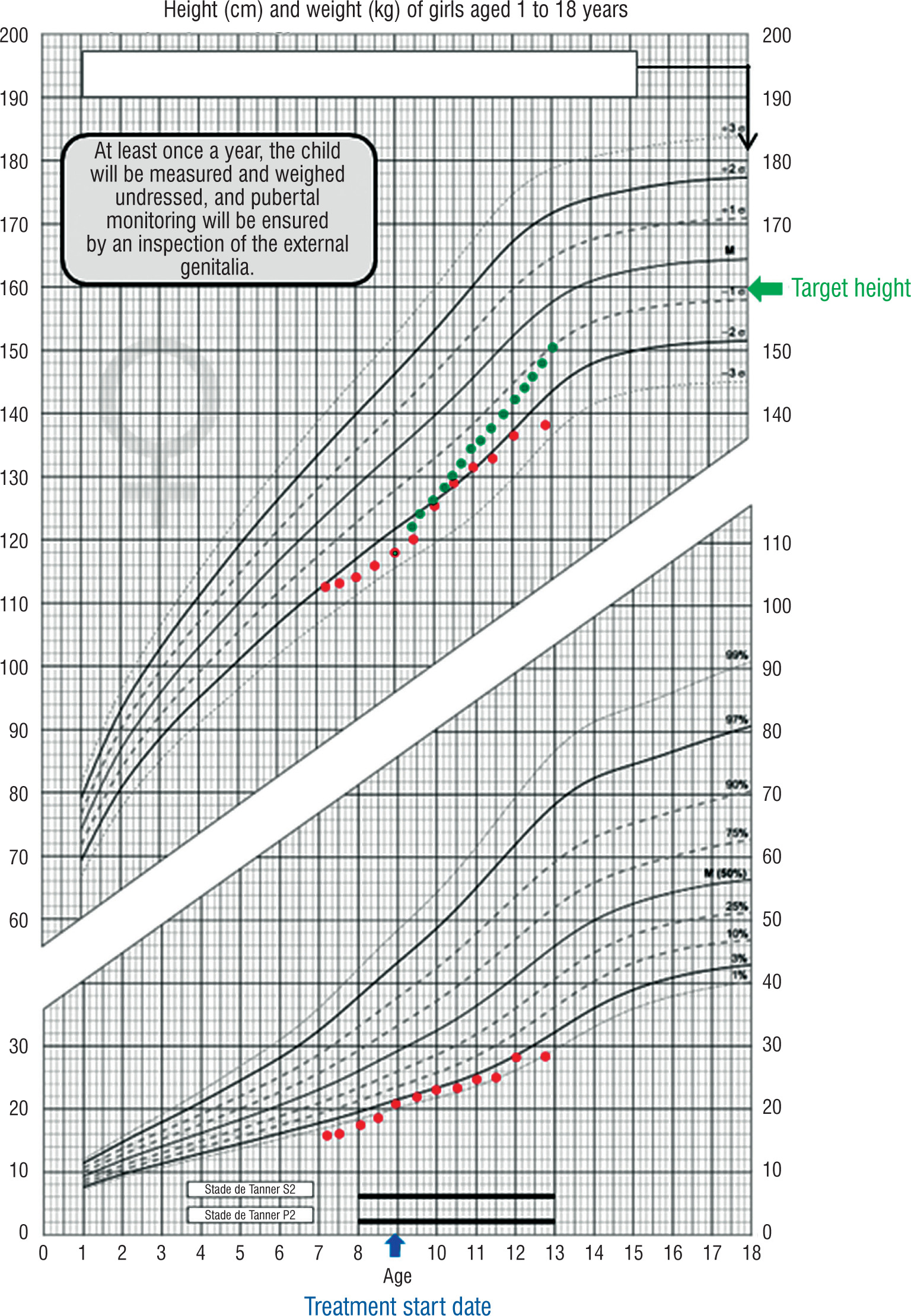
Despite four years of properly administered substitutive treatment with satisfactory therapeutic compliance, the stature response remained suboptimal with 5–6 cm/year growth velocity, below the expected 7–8 cm/year under hormonal substitutive therapy (Figure I). This insufficient clinical response contrasted with serum IGF-1 concentration normalization (241.7 ng/ml; age 10 normal range: 112–398 ng/ml), indicating biochemical-clinical discordance. This suboptimal response, while not unexpected in the context of emerging syndromic features, prompted a thorough diagnostic reevaluation.
Figure 1
Growth response comparison in Kleefstra-2 syndrome
Growth chart demonstrating suboptimal treatment response in Kleefstra-2 syndrome. Red dots: actual height progression in our patient under growth hormone therapy (initiated at 9 years, blue arrow). Green dots: expected growth response in typical idiopathic growth hormone deficiency. Despite 4 years of growth hormone treatment with IGF-1 normalization, growth velocity remained 5–6 cm/year vs. expected 7–8 cm/year, illustrating treatment resistance in Kleefstra-2 syndrome
GH – growth hormone; IGF-1 – insulin-like growth factor-1
Anthropometric examination revealed significant stature-weight delay at age 12 years and 10 months with height 138 cm (–2.5 SD), weight 28 kg (–3 SD), and a preserved body mass index (BMI) at 14.7 kg/m2 (–0.5 SD).
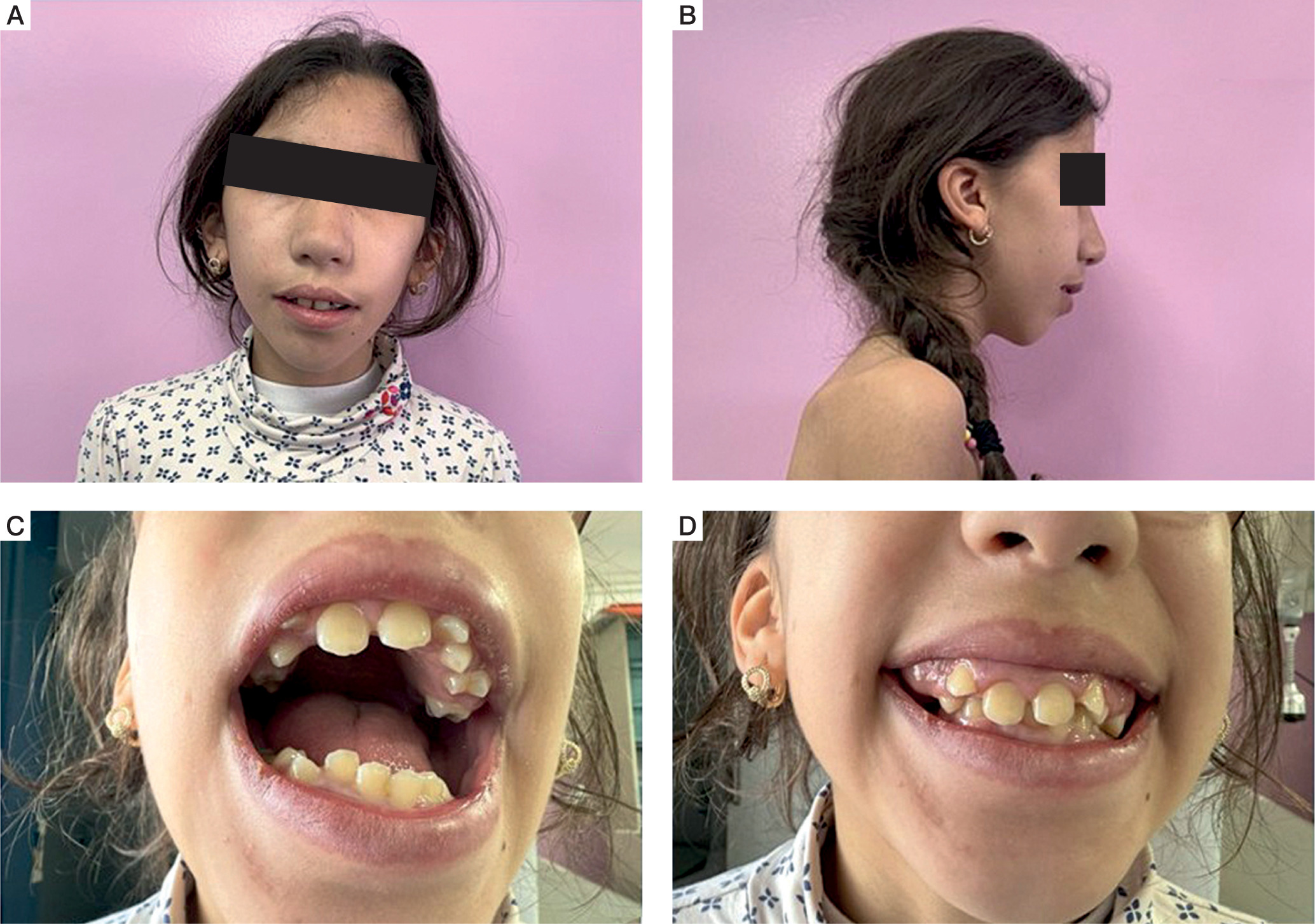
Morphological examination revealed dysmorphic facies characterized by triangular face shape, prominent and enlarged forehead, hypertelorism, cervical brevity, and maxillo-mandibular dysmorphia associated with dental malpositions and gingival hyperplasia (Figure 2). Neurological examination revealed moderate axial hypotonia. Neurocognitively, the patient presented globally satisfactory oral language development but with marked bradypsychia in response times, and severely impaired learning capacities requiring specialized educational management and speech therapy support.
Figure 2
Facial dysmorphic features in Kleefstra-2 syndrome. Clinical photographs showing characteristic dysmorphic features: A) frontal view demonstrating triangular face, broad forehead, and hypertelorism; B) profile view showing cervical brevity; C, D) intraoral examination revealing gingival hyperplasia and dental malpositions. Written parental consent was obtained for publication
Source: From the authors' archive. Publication with the consent of the parent/guardian
Hormonal assessment was normal, with normal thyroid, adrenal, and gonadal function. Bone age remained significantly delayed (10.5 years for chronological age 12.5 years). Metabolic analyses and standard karyotype were normal.
Facing this unexplained therapeutic resistance, associated with dysmorphic traits and neurodevelopmental delay, several differential diagnoses were considered, including Laron syndrome. However, the patient did not meet the diagnostic criteria for this syndrome, which requires severely reduced IGF-1 levels (typically < 20 ng/ml) and elevated GH levels. Moreover, the paradoxical IGF-1 normalization under treatment was incompatible with classic GH resistance and oriented toward alternative pathophysiological mechanisms.
Whole exome sequencing genetic analysis of the patient revealed heterozygous KMT2C gene mutation: c.7444_7445 insCC(p.Phe2482Serfs*34). This variation, not reported in variant databases, corresponds to two cytosine insertions causing a reading frame shift and presence of a premature stop codon at amino acid 2482. According to American College of Medical Genetics and Genomics (ACMG) classification criteria, this mutation was classified as likely pathogenic and confirmed to be de novo, excluding hereditary transmission.
These results established the diagnosis of Kleefstra-2 syndrome, unifying all observed clinical manifestations: refractory short stature, characteristic facial dysmorphia, and neurodevelopmental disorders. Kleefstra-2 syndrome diagnosis allowed reconsideration of the therapeutic strategy. Absence of significant stature gain after four years of growth hormone treatment (5–6 cm/year versus expected 7–8 cm/year), despite IGF-1 normalization, suggests post-receptor resistance to IGF-1 effects, likely related to epigenetic dysfunctions induced by KMT2C mutation. Growth hormone was progressively discontinued with clinical and biological tolerance monitoring. Management was reoriented toward the multidisciplinary approach adapted to Kleefstra-2 syndrome, including speech therapy follow-up for language disorders, psychomotor rehabilitation for hypotonia, orthodontic support for dental malpositions, and family genetic counseling.
Discussion
Our observation perfectly illustrates the diagnostic challenges of short stature associated with neurodevelopmental abnormalities. Insulin-like growth factor-1 normalization under growth hormone without proportional clinical improvement, in the context of facial dysmorphia and developmental disorders, suggests a complex genetic syndrome rather than isolated endocrine deficiency.
Facing this atypical clinical presentation, three main differential diagnoses should be considered. Growth hormone insensitivity syndromes, including Laron syndrome and JAK/STAT pathway mutations, typically present elevated GH levels with collapsed IGF-1 – the opposite profile to our patient [4]. Genetic multiple pituitary deficiencies usually accompany structural magnetic resonance imaging abnormalities and associated hormonal deficits, elements absent here [5]. Epigenetic syndromes, including Kleefstra-2 syndrome, represent an emerging category where growth retardation integrates into complex malformative presentation [6].
Recent observations suggest that endocrine abnormalities could be underdiagnosed in epigenetic machinery disorders. A 2019 systematic review showed that growth disturbances and hypothalamic-pituitary dysfunctions are frequent in these syndromes but rarely explored thoroughly [7]. These data emphasize the importance of systematic endocrinological evaluation in Kleefstra-2 syndrome.
Table I presents the distinguishing features between our case and classic GH deficiency, highlighting early warning signs: neurodevelopmental delay, facial dysmorphia, and paradoxical treatment resistance.
Table I
Differential characteristics between isolated GH deficiency, Laron syndrome, and Kleefstra-2 syndrome
[i] GH – growth hormone; IGF-1 – insulin-like growth factor-1; GHR – growth hormone receptor; KMT2C – lysine methyltransferase 2C Key diagnostic criteria: Biochemical-clinical discordance: IGF-1 normalization without adequate growth response (< 7 cm/year). Neurodevelopmental delay: language delay, intellectual disability. Characteristic dysmorphism: triangular face, broad forehead, hypertelorism.
Our observation brings three major contributions to Kleefstra-2 syndrome literature. First, we document the first growth hormone deficiency case with comprehensive hormonal evaluation including pathological stimulation tests. It should be acknowledged that GH stimulation tests have limited reproducibility and are not definitive for GH deficiency diagnosis, particularly in syndromic contexts where multiple factors can influence hormonal responses. Second, we describe unique therapeutic resistance despite IGF-1 normalization. Third, we identify a novel frameshift mutation (c.7444_7445insCC).
This mutation causes a premature stop codon, altering H3K4me1 methyltransferase function [8]. The resulting epigenetic perturbations probably compromise the transcriptional response to growth factors, explaining the ineffectiveness of treatment despite biochemical correction. This discovery enriches the syndrome mutational spectrum and illuminates interactions between epigenetic regulation and growth.
Literature analysis reveals that 53% of Kleefstra-2 patients present short stature, yet hormonal investigations remain insufficient [2]. Yang et al. reported severe cases without somatotropic axis evaluation [9]. The Rots et al. cohort mentions six patients treated with growth hormone with “apparently good results” without quantitative data [2]. Our observation constitutes the first complete endocrinological characterization with documentation of therapeutic resistance.
Distinction between Kleefstra-1 and Kleefstra-2 syndromes is crucial: type 2 presents less severe neurodevelopmental involvement, more autistic disorders, and 15% inherited forms [2]. This phenotypic variability could influence endocrine manifestations and the therapeutic response.
Facing ineffectiveness after four years of treatment and considering the economic constraints, we decided to discontinue progressive growth hormone. Management was reoriented toward a multidisciplinary approach including neuropediatric follow-up, speech therapy, and genetic counseling [10].
This experience advocates systematic endocrinological evaluation in Kleefstra-2 syndrome. Children with syndromic short stature and dysmorphic features rarely respond optimally to growth hormone treatment, as demonstrated in our case. Early recognition of the syndromic context would avoid costly and potentially ineffective therapeutics, guiding the implementation of multidisciplinary management from the time of diagnosis.
Conclusions
We report the first Kleefstra-2 syndrome case associated with growth hormone deficiency showing an abnormal substitutive treatment response. This observation expands the phenotypic spectrum of this rare syndrome and emphasizes the importance of considering genetic diagnoses in evaluating short stature refractory to conventional treatments. Additional studies are necessary to understand KMT2C and somatotropic axis interactions, which could open new therapeutic perspectives for patients with this syndrome. Multidisciplinary follow-up is essential to optimize these complex patients’ management.









