
Introduction
Precocious puberty refers to the appearance of secondary sex characteristics before the age of 8 years in girls and 9 years in boys. Clinically, there are two types of precocious puberty – gonadotropin-dependent (central) and gonadotropin-independent (peripheral) – according to the activation of the hypothalamic-pituitary-gonadal axis [1, 2]. Central precocious puberty (CPP) incidence is 10- to 20-fold higher in girls [3]. About 80–90% of CPP cases in girls are idiopathic, while 50–70% of boys have identifiable pathological changes [1, 2]. The etiology is miscellaneous, including congenital and acquired causes. Mutations encoding kisspeptin (KISS1), kisspeptin receptor (KISS1R), makorin ring finger protein 3 (MKRN3), and delta-like noncanonical notch ligand 1 (DLK1) have been identified in familial disease [4, 5]. Hypothalamic hamartoma (HH) is one the most frequently observed congenital causes of CPP with central nervous system (CNS) abnormalities [4]. Among other congenital causes, the literature mentions suprasellar arachnoid cysts, hydrocephalus, neurofibromatosis type 1 (NF1) causing optic pathway tumors, tuberous sclerosis complex (TSC), septo-optic dysplasia (SOD), Chiari II malformation, and myelomeningocele [4–8]. Syndromic disorders are also associated with CPP (Sturge-Weber, Prader-Willi syndrome, Silver-Russell, Rett, and Temple syndromes) [4].
Both CNS tumors – astrocytomas, ependymomas, pinealomas, and optic and hypothalamic gliomas – and their therapy may lead to CPP [8, 9]. Other acquired etiologies of CPP include granulomatous disease, previous sex steroid excess, and hypothalamic inflammation. CPP may be a rare complication of CNS irritation, but when it occurs, it is commonly associated with growth hormone deficiency [10, 11]. Inappropriate gonadotropin secretion varies depending on the dose of irradiation received [12]. Studies have shown that lower doses (< 30 Gy) of irradiation in children contributed to precocious puberty. However, higher doses (> 30 Gy) caused hormonal deficiencies [12–14].
Craniopharyngiomas (CPs) are rare and benign, slow-growing CNS tumors, among primary CNS tumors, as the second most common neoplasms in children and the most frequent solid tumors [15]. At the follow-up after treatment, patients with CP present with anterior panhypopituitarism to a similar extent between those treated with surgery only (78%) and those who underwent surgery and radiotherapy (86%) (p = 0.23). More than 80% of patients suffer from at least one hormone-axis insufficiency [16]. Nevertheless, CPs can also rarely present with precocious puberty, at a frequency of about 2%, but only a few cases are described in the literature [17–19]. Therefore, before proceeding to treatment, current guidelines recommend meticulous analysis of hormonal pituitary function with basal measurements and dynamic tests [17].
This paper introduces the case of a 6.5-year-old girl with craniopharyngioma, who, after a surgical procedure and head irradiation with a dose of 54 Gy, developed CPP. Despite growth hormone deficiency, she continued to grow within the normal range over 7 years following the CP diagnosis. Therefore, this article aims to discuss the true clinical picture of the disease after CP therapy and approaches to achieving optimal care.
Methods
The PubMed and Google Scholar databases were searched for relevant sources during the period from January 2024 to September 2025 to identify studies for inclusion in this review paper. Only studies published in English or Polish and based on human research were considered eligible. The review focused on studies involving the pediatric population. The selection process included original articles, review papers, meta-analyses, case studies, book chapters, and current guidelines and recommendations. Literature was assessed using the following keywords: craniopharyngioma, head irradiation, suprasellar tumors, precocious puberty, and growth without growth hormone. Abstracts of the identified studies were first reviewed. Later, full-text articles were obtained if they met the eligibility criteria, and then ultimately either accepted or declined for the purpose of this review.
Bioethical standards
Written informed consent was obtained from the participant’s parents to publish the details of this medical case and accompanying images.
According to the Polish law and Good Clinical Practice regulations this research does not require approval of the Bioethics Committee at Poznan University of Medical Sciences (decision number: KB - 681/25).
Case report
The 6.5-year-old girl, who was born prematurely from a twin pregnancy by cesarean section at 31 weeks of gestational age (WGA), with a body weight of 1,450 g and a 9-point Apgar score, was admitted to the endocrinology department of the university hospital due to precocious breast development. Her past medical history revealed that at the age of 4 years, she was diagnosed at the neurology clinic due to paroxysmal episodes of headache, which had presented since the age of 3 years. The headaches, which repeated 2 to 3 times a month, were localized in the occipital area and accompanied by vomiting. Afterwards, the girl fell asleep and subsequently woke up without any complaints. Until then, her developmental milestones had been achieved on time. While the neurological evaluation showed no abnormalities, binocular hyperopia and astigmatism were found during the ophthalmologic examination. Further diagnostic procedures included serological tests for Toxoplasma gondii and Borrelia burgdorferi, abdominal ultrasound examination, and electroencephalography, which were normal. Cerebral magnetic resonance imaging (MRI) showed a large intraventricular colloid cyst of the third ventricle, of diameters 39 × 23 × 25 mm (width × length × height), and features of hydrocephalus in the lateral ventricles.
The surgical treatment (craniotomy) was performed in the department of neurosurgery, where the tumor, macroscopically suspected to be CP, was partially resected. The histopathological examination revealed adamantinomatous CP ameloblastic type (World Health Organization grade 1 – G1), surrounded by piloid gliosis with abundant Rosenthal fibers. Radiotherapy with a total irradiation dose of 54 Gy was indicated. However, its course was interrupted by staphylococcal sepsis and was finally completed at 4 years and 4 months.
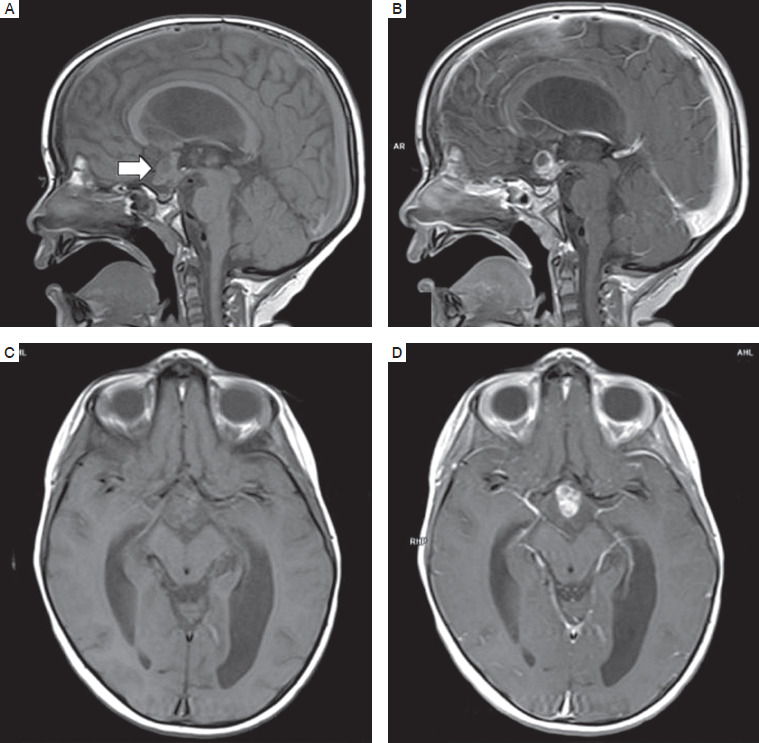
Subsequently, follow-up MRI of the head revealed remnants of the tumor in the form of a multicystic lesion in the suprasellar cistern, dorsally compressing the optic chiasm, slightly constricting the third ventricle, of diameters 18 × 14 × 20 mm (Fig. 1). MRI examinations were repeated regularly every 3 months, showing slow regression of the tumor mass reaching the diameters 7.9 × 7.5 × 12.5 mm. During a follow-up visit 2 years after she completed the therapy, the oncologist noted an advanced pubertal stage.
Figure 1
MRI of the brain in the reported patient after completing oncologic therapy (A. T1 sequence in the sagittal plane; B. T1c sequence in the sagittal plane; C. T1 sequence in the axial plane; D. T1c sequence in the axial plane)
In the endocrinology department, the patient’s height was 121 cm (50th–75th percentiles for age), and body weight was 29.5 kg (90th–97th percentiles for age). She presented with deposition of adipose tissue at her trunk and a nonpalpable thyroid gland, and her sexual development on the Tanner scale was as follows: Thelarche (Th) 3, Axillarche (Ax) 1, Pubarche (Pub) 1/2. The bone age assessed based on the Greulich & Pyle method was 10 years, and the predicted final height determined by bone age was 146.1 cm, while the parental estimated height prognosis was 167 cm (50th–75th percentiles).
The laboratory functional workup of the pituitary axis was carried out. The results of the hormonal workup in the patient are displayed in Table I.
Table I
Levels of hormonal parameters at diagnostic work-up
* According to the Polish recommendations.
Deviations from the norm are highlighted in bold.
ACTH - adrenocorticotropic hormone; AFP - alpha-fetoprotein;
b-HCG - human chorionic gonadotropin; DHEA-S - dehydroepiandrosterone sulfate; FSH - follicle-stimulating hormone; fT3 - free triiodothyronine; fT4 - free thyroxine; GH - growth hormone; IGF-1 - insulinlike growth factor-1; LH - luteinizing hormone; LH-RH - luteinizing hormone-releasing hormone; TSH - thyroid-stimulating hormone
The ultrasound examination revealed a normal picture of the thyroid gland. The girl was also evaluated by a gynecologist. An ultrasound revealed advanced pubertal development of the genital system (uterus size of 40 mm length, anteroposterior and transverse diameter of a uterus body of 13 and 25 mm, respectively, cervix 14 mm, endometrial thickness 1 mm, size of the right ovary 20 × 12 × 14 mm, diameter of the largest follicle 12 mm, left ovary dimensions 15 × 11 × 10 mm, and diameter of the largest follicle 9 mm).
Based on the personal history, including the suprasellar tumor and the radiotherapy of the CNS, the clinical presentation with Tanner stage (Th3), the laboratory hormonal findings, accelerated bone age (10 years), and the ultrasound features of pubertal development of the uterus and ovaries, central, gonadotropin-releasing hormone (GnRH)-dependent precocious puberty was diagnosed. Therapy with long-acting GnRH analog triptorelin was initiated. After 3 months of treatment, the stimulation test with luteinizing hormone-releasing hormone (LH-RH) was performed; it demonstrated suppressed levels of luteinizing hormone (LH) = 0.29 mIU/ml (peak value) and follicle-stimulating hormone (FSH) = 2.2 mIU/ml (peak value), LH/FSH ratio 0.13, and an undetectable serum estradiol level (below 10 pg/ml).
At the most recent follow-up visit, more than 4 years after the diagnosis of CPP, the patient was 11 years and 1 month old. She was continuing the therapy with triptorelin. Her height was 149 cm (50th–75th percentiles), and her weight was 54.5 kg (90th–97th percentiles); thus, her body mass index (BMI) (24.5, 97th percentile) indicates that she is obese. Still, the pubertal status is arrested (Th3, Pub2). The bone age was determined to be 11 years and 6 months.
Nevertheless, an immense improvement in predicted adult height was noted, as her height was 158 cm (Fig. 2). The results of the laboratory work-up are shown in Table II.
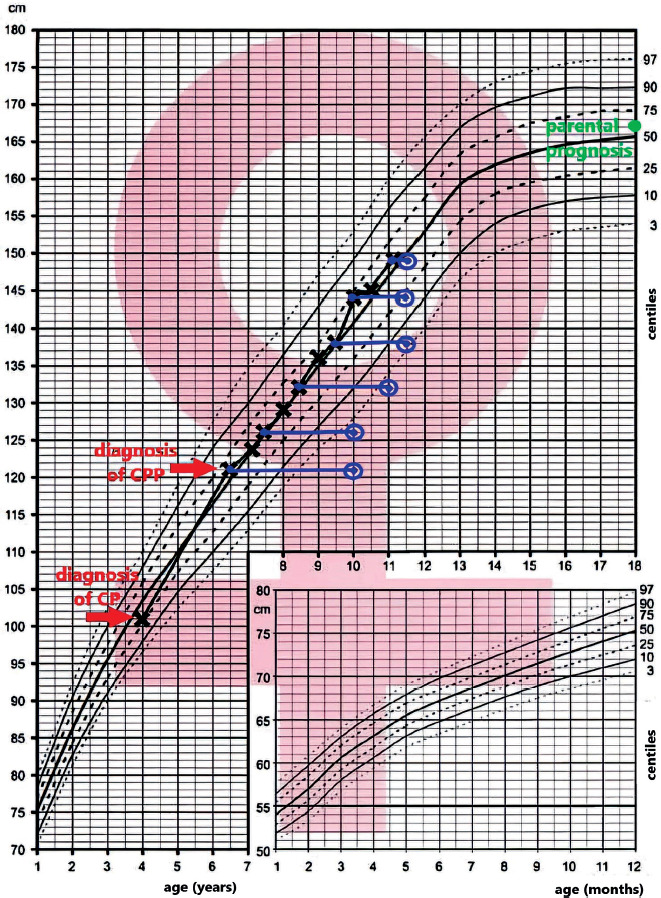
Table II
Current levels of hormones in the patient at age 11 years
[i] Deviations from the norm are highlighted in bold.
ACTH - adrenocorticotropic hormone; DHEA-S - dehydroepiandrosterone sulfate; FSH - follicle-stimulating hormone; fT3 - free triiodothyronine; fT4 - free thyroxine; HDL - high-density lipoprotein; IGF-1 - insulin-like growth factor-1; IGFBP3 - insulin-like growth factor-binding protein 3; LDL - low-density lipoprotein; LH - luteinizing hormone; LH-RH - luteinizing hormone-releasing hormone; TSH - thyroidstimulating hormone
In follow-up MRI, the pituitary was normally located and symmetrical, 3.5 mm in height, 9 mm in width, and 6.5 mm in anterior-posterior dimension. In T1-dependent images without contrast medium, the signal appeared normal. Along the upper pituitary stalk, a tumor was visible, affecting the shape of the optic chiasm. The image of brain structures remained unchanged compared to the imaging in the previous years (Fig. 3).
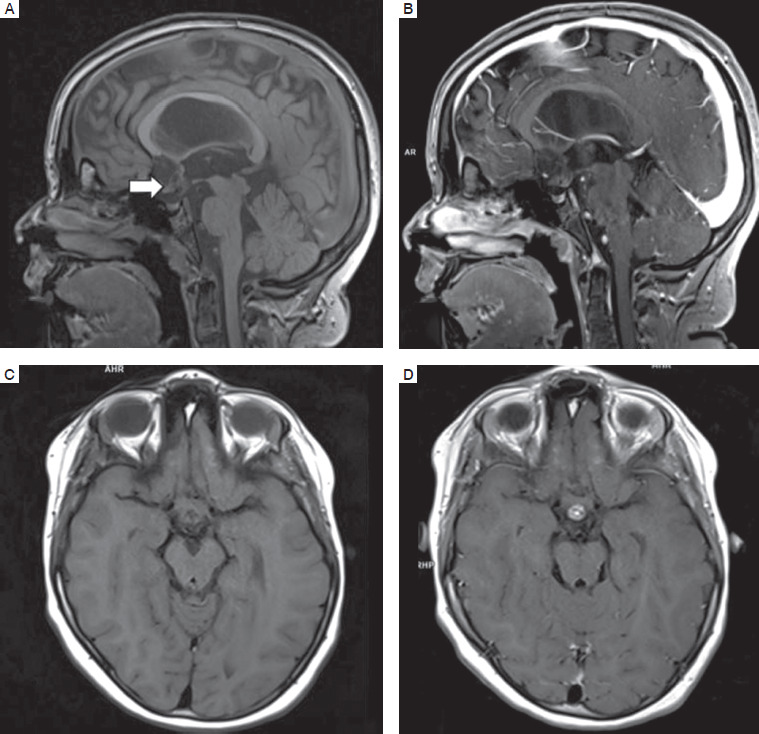
Discussion
CPs account for 2–5% of CNS tumors [20], and among them, 30–50% of all cases are diagnosed in children, most frequently at the age between 5 and 14 years, but rarely in neonates or prenatally [14, 20–24]. They are among the most frequent CNS tumors, alongside astrocytomas, medulloblastomas, and ependymomas [25, 26], and in children, they make up 5–10% of all intracranial tumors [27]. No gender-related diversity has been observed in population-based research [28]. Notably, the reported patient presented with initial symptoms of the disease at 3 years of age, and the diagnosis was established at 4 years, so below the age of the predominant frequency. CPs are benign, slow-growing CNS tumors, most frequently localized in the suprasellar region with an intrasellar component, less frequently in the purely intrasellar region. They tend to compress or invade the adjacent structures, such as the optic chiasm, hypothalamic-pituitary pathway, or third ventricle [22, 26, 27, 29]. The structure of CPs can be cystic or mixed cystic-solid, with calcifications being present in nearly 90% of cases [27]. It is worth noting that in the presented patient, the tumor was localized in the suprasellar region and the third ventricle, and its structure was cystic-solid.
CPs are classified into two categories: adamantinomatous and papillary. The adamantinomatous type is most prevalent among children, as is the case with the described patient [26]. It probably originates from the epithelial remnants of the craniopharyngeal duct or Rathke’s pouch [27]. It is associated with an activating mutation in the third exon of the CTNN1 gene encoding β-catenin [30]. Its effect is the nuclear accumulation of β-catenin and constitutive activation of the Wnt pathway. The papillary type is characteristic for adults and is formed due to metaplasia of residual squamous epithelial cells of the anterior pituitary and infundibulum. The BRAF-V600E mutation is found in 95% of papillary-type patients [30].
When the tumor resides in the third ventricle, acute hydrocephalus symptoms frequently manifest, prompting an early diagnosis. Predominantly, they occur up to 2 weeks before diagnosis. In the presented clinical case, headaches accompanied by vomiting had been observed for a year before the diagnosis of CP. Visual defects also occurred but had not initially raised oncological concerns due to the low frequency and severity of occurrence.
Chronic clinical symptoms of CP result from the location of the tumor near the vital structures such as the hypothalamus, pituitary gland, optic nerves, and cerebral arteries [20]. Direct damage to, or compression of typical structures can lead to various endocrine abnormalities. Growth failure caused by either hypothyroidism or GHD is the most common presentation in children. In about one-third of the patients, weight gain is observed due to hypothalamic obesity, and the same number of patients present with growth failure and short stature [17]. As many as 54% to 92% of the patients have at least one pituitary hormone deficiency at the time of CP diagnosis. Pituitary insufficiency occurs more often in childhood-onset than in adult-onset CPs [31]. Some CP patients are diagnosed with diabetes insipidus (DI) preoperatively, and about 10% manifest pubertal delay or arrest [17, 28, 32, 33].
The presented girl experienced GHD at an unknown time of onset, which did not encompass growth deceleration either before or after CP diagnosis, and was subsequently masked by CPP. Visual disturbances that correlate with the tumor often include decreased visual acuity, visual field limitation, optic disc swelling, optic nerve atrophy, double vision, and strabismus. Neurological disorders include consciousness disorders, hemiparesis, and balance disorders [20, 26, 27, 31, 34]. The described patient was initially diagnosed only with visual acuity disorders and astigmatism, but the last examination revealed visual field defects.
Treatment of CPs involves surgery through the sphenoid sinus or craniotomy. However, gross total resection is often impossible due to closeness to critical structures such as optic pathways, the circle of Willis, and the hypothalamus [35]. Previous studies also stated that complete resections were often associated with increased mortality due to hypothalamic dysfunction and neurological functions [36, 37]. The latest guidance on managing pediatric CPs recommends moderate strength and not proceeding with the complete resection, where there is clear evidence of hypothalamic involvement [17]. The hypothalamus-sparing surgical (HSS) strategy results in a better quality of life (QoL) and prevents negative consequences, especially the development of obesity. Since this approach may be related to higher risk of relapse and progression, radiotherapy is widely used to eliminate the residual tumor [36, 37]. Currently, recommended radiation techniques are photon-based radiation therapy (XRT) and proton beam therapy (PBT) [17]. Both are associated with the same doses of radiation, and comparable survival rates and extent of endocrinological sequelae [35–37].
Nevertheless, PBT focuses more on the treatment area, thus limiting the radiation the surrounding tissues receive, and reducing their damage and resulting complications [36]. In the presented clinical case, the initial size of the tumor was 39 × 23 × 25 mm, and after radiotherapy completion, the residual tumor mass was 18 × 14 × 20 mm. Over the next 36 months, it gradually decreased to 7.9 × 7.5 × 12.5 mm.
After surgery and radiotherapy, patients often require endocrine care for hypothalamic-pituitary disorders. Growth deficiency resulting from the presence of CP is one of the indications for recombinant growth hormone (rGH) therapy. In children with CP, there are isolated reports in the literature describing normal growth despite a measurable deficiency in growth hormone, likely due to the effects of leptin or insulin [38]. The presence of receptors for growth hormone, IGF-1, somatoliberin, estrogens, progesterone, and leptin was found in the CP tissue [39]. Their action may be important in developing and treating tumors, affecting the safety of replacement therapy with rGH. Especially when there is a residual tumor mass, the decision to initiate treatment should be made carefully due to the possibility of stimulating the re-growth of CP. In the outlined case report, the residual tumor mass presented with the dimensions of 7.9 × 7.5 × 12.5 mm; therefore, the implementation of rGH therapy seems to be controversial. However, reports in currently available literature suggest that this treatment does not contribute to tumor recurrence [40–42]. Treatment with rGH after completing oncological therapy is usually indicated following careful discussion with the patient and, if appropriate, their family, as well as consultation with the treating oncologist or neurosurgeon. Guidelines from the Endocrine Society and the Pediatric Endocrine Society suggest waiting 12 months after the completion of cancer treatment before initiating rGH therapy [43, 44]. In children with CPs, which are considered benign tumors, growth hormone (GH) therapy may be safely started as early as 0.7 years after diagnosis [43]. At present, the initiation of rGH is not recommended after oncological treatment with tyrosine kinase inhibitors (TKI). The dosing of rGH is similar to that used for the treatment of GHD in children who are not cancer survivors. Physicians should ensure that IGF-1 levels are maintained appropriately for the patient’s sex, age, and pubertal status [43–45]. The research did not demonstrate an increased risk of CP recurrence following initial neurosurgery in patients treated with rGH [45]. In addition, a recent meta-analysis suggested that recurrence rates of CP were reduced in children receiving rGH [41]. In the presented clinical case, the most probable cause of precocious puberty is previous irradiation of the CNS, along with the other coexisting risk factors. Cancer survivors who underwent cranial irradiation at a dose as high as 18 Gy or more are at risk of developing CPP [46]. Additional risk factors include a history of hydrocephalus or tumors in the hypothalamus-pituitary region. Furthermore, among patients who experienced radiation therapy to the hypothalamic region, the risk is amplified by younger age at diagnosis and female sex, both of which align with the profile of our patient [7].
Studies have shown that lower doses (< 30 Gy) of irradiation in children contributed to precocious puberty in females but not in males, whereas higher doses (24–50 Gy) can induce precocious puberty in both sexes [12–14]. The sexual features of puberty emerged, on average, at 8.5 years of age [12, 13]. The underlying pathophysiological mechanism relies on the damage caused to GABAergic neurons in the cerebellum and cerebral cortex, which can lead to a decrease in the concentration of the neurotransmitter GABA and a loss of its function as an inhibitor of the pulse generator [7]. Furthermore, Xu et al. found, in a mouse model, that cranial radiation can induce overactivation of the p53 pathway and simultaneously inhibit the Hippo pathway, thus leading to enhanced cell apoptosis, decreased cell proliferation, and subsequent pituitary injury [31, 47].
On the other hand, studies have demonstrated that high-dose therapies are more likely to cause gonadotropin deficiencies and delay the onset of puberty [46, 48–50]. Radiation doses of 30–50 Gy were associated with a 20–60% incidence of long-term gonadotropin deficiency [49]. Other hormonal deficiencies besides LH and FSH include GH, thyroid-stimulating hormone (TSH), and adrenocorticotropic hormone (ACTH) shortages. At the same time, GH tends to be the most susceptible, and the other two are relatively resistant to the effects of radiation [12–14]. The deficiencies resulting from irradiation seem irreversible and progressive [13]. Chemaitilly et al. [49], after analysis of a large cohort of patients with childhood-onset cancers who underwent cranial radiotherapy, stated that at 40 years from cancer diagnosis, the most frequently deficient was GH (with an estimated cumulative incidence of 72.4%), then LH and FSH (24.4%), followed by TSH (11.6%) and ACTH (5.2%). The authors found a significant association between gonadotropin deficiency and male sex, as well as white race [49]. The patient in question underwent radiotherapy with a dose of 54 Gy at the age of 4, so she received a high dose of radiation at a young age. Just 2 years after treatment, she experienced the first symptoms of premature puberty.
It has been shown that in patients who have both the diagnosis of a brain tumor and premature puberty, precocious puberty occurs in as many as 60% of cases following prior radiotherapy. In 5% of cases, precocious puberty coexisted with the diagnosis of the tumor; in 17.5% of cases, it appeared after the diagnosis of the tumor but before radiotherapy, and in 17.5% of cases, it emerged in patients who did not undergo radiotherapy [10].
Premature puberty, as a sequel of radiotherapy for CP, may coexist with hypopituitarism in terms of somatotropin secretion, which has a double negative impact on the final target height. It has been shown that in the case of early diagnosis and immediate initiation of treatment for premature puberty, its impact on final height is insignificant [10, 50]. In the described patient, a severe growth hormone deficiency was demonstrated in the nocturnal growth hormone secretion test and after stimulation with glucagon. Initiation of rGH therapy to improve the patient’s final height could be considered. However, the child’s height remains above the 50th percentile on the growth chart, with only slightly accelerated bone age at the recent follow-up.
Nevertheless, the patient still receives puberty-delaying therapy with triptorelin to achieve a greater final height. Yet her appropriate pace of growth is baffling. Considering that she has developed obesity alongside dyslipidemia, obesity may be a key factor contributing to her growth processes through the actions of insulin and leptin, thereby sustaining her unexpectedly adequate growth velocity. On the other hand, the metabolic dose of rGH, because of metabolic disturbances, may be beneficial for her.
Summary
In the presented clinical case, the patient experienced symptoms of increased intracranial pressure (ICP), including headaches and vomiting, for more than a year before diagnosis. She was initially diagnosed with ophthalmological disorders, including impaired visual acuity and, subsequently, visual field defects. Ultimately, a diagnosis of craniopharyngioma (CP) was established. Partial tumor resection was performed via craniotomy, followed by radiotherapy with a total dose of 54 Gy.
During the first 2 years of follow-up after completion of oncological treatment, serial MRI examinations demonstrated a gradual reduction in the residual tumor size. Two years after radiotherapy, the patient developed precocious puberty, with breast enlargement as the first clinical sign. While this could theoretically be related to the brain tumor itself and the associated ICP, the significant time interval suggests that premature puberty was most likely a consequence of high-dose cranial irradiation at a very young age.
Given the risk of compromised final adult height due to premature puberty, advanced bone age, growth hormone deficiency, and potential metabolic complications such as dyslipidemia and elevated BMI, rGH therapy was considered. However, in light of the patient’s normal growth velocity, current height above the 50th percentile with concordant bone age, and the potential risk posed by residual tumor tissue, a decision was made to defer initiation of rGH therapy.









