
Introduction
Congenital hyperinsulinism represents the main cause of persistent hypoglycemia in infants, affecting 1 birth in 25,000 to 50,000 [1]. This condition, characterized by inappropriate insulin secretion, shows increasing genetic heterogeneity, with more than 12 genes identified to date [2]. The identified mutations mainly concern proteins involved in the regulation of insulin secretion by pancreatic β cells.
Among these genes, HADH, coding for the enzyme short-chain L-3-hydroxyacyl-CoA dehydrogenase, is particularly interesting. Mutations of this gene disrupt fatty acid metabolism and represent an established cause of autosomal recessive congenital hyperinsulinism [2]. Loss of HADH function leads to accumulation of short-chain fatty acids and altered ATP production, resulting in inappropriate insulin secretion [3]. Patients with isolated HADH mutations typically present with moderate to severe phenotypes, with a variable age of onset, often greater than 3 months [3].
In parallel, abnormalities in the GHSR gene, coding for the ghrelin receptor, have recently been implicated in glucose homeostasis regulation. Ghrelin plays a crucial role in glycemic counter-regulation and suppression of insulin secretion [4]. Isolated GHSR mutations are generally associated with mild to moderate metabolic disturbances, with moderate growth delay (–2 to –3 SD) and rare neurological manifestations [5].
The association of HADH and GHSR mutations has never been reported in the literature. This combination could theoretically lead to pathological interactions between lipid metabolic drivers and hormonal signaling, potentiating their respective effects on glucose homeostasis. The metabolic pathways involved converge at the level of cellular energy regulation and hormonal signaling [5].
We describe here the first case of severe congenital hyperinsulinism combining novel mutations in HADH and GHSR genes, illustrating a new digenic mechanism in the pathogenesis of this disease. This observation allows us to explore the interactions between fatty acid metabolism and hormonal signaling in glucose homeostasis regulation, opening new therapeutic perspectives for complex forms of congenital hyperinsulinism. Furthermore, this case highlights the significant challenges in managing complex genetic forms of CHI in resource-limited settings, where access to specialized medications, continuous glucose monitoring systems, and advanced diagnostic tools is often restricted. These limitations necessitate innovative adaptations of standard treatment protocols and underscore the need for context-specific management strategies in regions where healthcare resources are constrained.
Case report
We report the case of a male patient, currently 7 years old, who was initially admitted at 24 days of life for severe hypoglycemic seizures. Written informed consent was obtained from the parents for the publication of this case report and any accompanying images. Born at term from an uncomplicated pregnancy to non-consanguineous parents, he had no family history of hyperinsulinism or metabolic disorders. Birth parameters were normal with a weight of 3,300 g (0 SD), length of 50 cm (0 SD), and head circumference of 35 cm (0 SD).
Initial symptoms were characterized by generalized tonic-clonic seizures occurring three times daily, lasting 2–3 minutes, predominantly in the postprandial period. Initial examination revealed severe hypoglycemia at 0.3 mmol/l (N: 3.5–5.5 mmol/l), associated with inappropriate hyperinsulinemia at 10.31 µIU/ml (N: < 2.0 µIU/ml during hypoglycemia) and C-peptide at 2.64 µg/l (N: < 0.5 µg/l during hypoglycemia).
In-depth biochemical analysis revealed major metabolic disturbances. Comprehensive metabolic workup demonstrated global hyperaminoaciduria (23,190 µmol/l equivalent to 2,906 µmol/mmol, N: 475–2,152 µmol/mmol), with markedly elevated urinary glycine (1,379 µmol/mmol, N: 110–356 µmol/mmol) and plasma glycine (846 µmol/l, N: 224–324 µmol/l). The acylcarnitine profile showed increased free carnitine (65.12 µmol/l, N: 13–52 µmol/l) and acetylcarnitine (36.17 µmol/l, N: 16–35 µmol/l), along with elevated butyrylcarnitine. Persistent hyperammonemia (93 µmol/l, N: 16–60 µmol/l) and hyperlactatemia (3.51 mmol/l, N: 05–2.2 mmol/l) completed this picture, suggesting profound disruption of cellular energy metabolism.
Endocrine evaluation during hypoglycemia (0.3 mmol/l) revealed inadequate GH response (8.35 µIU/ml, N: > 20 µIU/ml) and low IGF-1 (27.8 ng/ml, N: 51–327 ng/ml), consistent with growth hormone deficiency.
Additional findings included elevated CPK (750 IU/l, N: < 190), moderately elevated TSH (5.3 mIU/l, N: 0.72–11 mIU/l), and low morning cortisol (146 nmol/l, N: 171–536 nmol/l). These alterations in both amino acid and acylcarnitine metabolism, combined with elevated ammonia and lactate levels, point to complex metabolic dysregulation affecting multiple pathways critical for energy homeostasis, consistent with the dual genetic alterations subsequently identified.
The neurological evolution rapidly deteriorated with a transformation of the epileptic profile. Initial tonic-clonic seizures evolved into infantile spasms at 12 months of age, confirming West syndrome with electroencephalography (EEG) showing characteristic hypsarrhythmia. At age 3, the clinical picture changed to Lennox-Gastaut syndrome, combining tonic seizures, atypical absences, and drop attacks, with EEG showing diffuse slow spike-waves. Psychomotor development remained stagnant, without achieving expected developmental milestones. The child never acquired complete head control, a sitting position, or walking ability. Growth delay progressively worsened, reaching –4 SD for both weight and height, associated with microcephaly at –2 SD.
Due to initial financial constraints, genetic analysis was delayed until age 7, when it became crucial for family planning and also because of the refractory nature of encephalopathy. Molecular investigation was performed using whole exome sequencing (WES), which identified a novel combination of mutations: a homozygous deletion in the HADH gene (5.25 kb, exons 3–4) detected through copy number variant (CNV) analysis (ExomeDepth software), and a heterozygous missense variant in the GHSR gene (c.611C>A, p.Ala204Glu) identified through single nucleotide variant (SNV) analysis (GenoSystem Variant Analysis software). Given the unprecedented nature of this mutation combination and its implications for future pregnancies, parental genetic testing was initiated to determine the inheritance patterns of both variants and provide appropriate genetic counseling for the family.
This combination had never been described in the literature. The involvement of the HADH gene in fatty acid metabolism and insulin regulation, combined with GHSR’s role in appetite regulation and glucose homeostasis, suggests a possible interaction explaining the severity of the clinical presentation.
Therapeutic management required a carefully adapted approach due to the local healthcare context in Morocco. Initial treatment at 24 days of life was based on the clinical presentation of severe hyperinsulinemia hypoglycemia. Management consisted of MCT-enriched formula (75 ml/2 h, specially formulated with protein, carbohydrates, MCT-rich/LCT-poor fats) and levocarnitine (100 mg/kg/day). Although diazoxide was initially prescribed following international recommendations for congenital hyperinsulinism, medication unavailability in Morocco and financial constraints prevented its acquisition. Glycemic monitoring targeted levels above 3.5 mmol/l, with monitoring frequency progressively adjusted from hourly checks during the initial phase to 4–6 pre-prandial checks daily in the current phase. After 6 months of age, uncooked cornstarch was progressively introduced (20 g/100 ml, with a total prepared volume of 400 ml) as nocturnal continuous feeding for 8–10 h. The infusion began at 20 ml/h and was adjusted throughout the night according to glycemic control measurements, allowing for personalized dosing to maintain target glucose levels. Treatment response was assessed through glycemic control, clinical symptoms, and neurological status. Despite these interventions, glycemic control remained suboptimal, reflecting the challenges of managing complex metabolic conditions in resource-limited settings. The genetic diagnosis at age 7, revealing the HADH and GHSR mutations, provided retrospective understanding of the treatment challenges faced throughout the patient’s course.
Currently, at age 7, the child presents with severe neurological impairment with significant dependence for daily activities. His follow-up is managed by a multidisciplinary team including a pediatric neurologist, endocrinologist, gastroenterologist, pulmonologist, and rehabilitation specialists. Recent investigations confirm the persistence of epileptic encephalopathy with progressive cortico-subcortical atrophy on brain magnetic resonance imaging (MRI). The clinical evolution and therapeutic management are summarized in Table I.
Table I
Clinical and therapeutic evolution from initial phase to 7 years of age
Discussion
This clinical observation presents several unique characteristics that enrich our understanding of severe congenital hyperinsulinism and its pathophysiological mechanisms.
The early onset and severity of initial manifestations constitute the first remarkable element. The onset of symptoms at 24 days of life, with profound hypoglycemia (0.3 mmol/l) associated with inappropriate hyperinsulinemia (10.31 µUI/ml), contrasts with the typically later onset of hyperinsulinism linked to isolated HADH mutations, usually diagnosed after 3 months of life [3]. This early presentation suggests a synergistic effect of combined HADH-GHSR mutations on neonatal glucose homeostasis.
The unfavorable growth evolution of our patient, reaching –4 SD for weight and height at 7 years, differs significantly from reported cases of isolated HADH mutations, where growth retardation rarely exceeds –2 SD [3, 6]. This divergence could be explained by the additional impact of GHSR mutation on the somatotropic axis, as ghrelin is an important growth hormone secretagogue.
The neurological evolution shows particular severity in our observation. The evolutionary sequence – neonatal seizures, West syndrome at 12 months, then Lennox-Gastaut syndrome at 3 years – represents a cascade of neurological events never described in cases of isolated congenital hyperinsulinism. This unfavorable progression [7], associated with marked pharmacoresistance, could result from the combined effect of early severe hypoglycemia, loss of ghrelin’s neuroprotective properties due to GHSR mutation, and complex metabolic disturbances linked to HADH mutation.
The therapeutic resistance observed in our patient is also notable. Despite intensive management combining specialized MCT-enriched formula (75 ml/2 h), levocarnitine (100 mg/kg/day), and nocturnal cornstarch (400 ml at 20 ml/h), glycemic control remained suboptimal, as evidenced by persistent pre-prandial glucose fluctuations and continued neurological manifestations. Local healthcare constraints, including unavailability of certain medications such as diazoxide, necessitated adaptation of standard therapeutic protocols. The genetic diagnosis at age 7 helped explain this therapeutic resistance, with the combination of HADH and GHSR mutations suggesting more complex pathophysiological mechanisms requiring innovative therapeutic approaches. This relative treatment resistance contrasts with the generally favorable response described in cases of isolated HADH mutations [3, 8], as detailed in Table II.
Table II
Comparison between our case and previously reported isolated HADH mutations cases
| Characteristics | Our case (HADH + GHSR) | Isolated HADH mutations [3, 6–8] |
|---|---|---|
| Age of onset | 24 days | > 3 months |
| Initial glucose level | 0.3 mmol/l | 1.2–2.8 mmol/l |
| Growth at 7 years | –4 SD | –1 to –2 SD |
| Epilepsy | West then Lennox-Gastaut | Rare/Absent |
| Treatment response | Partial | Generally good |
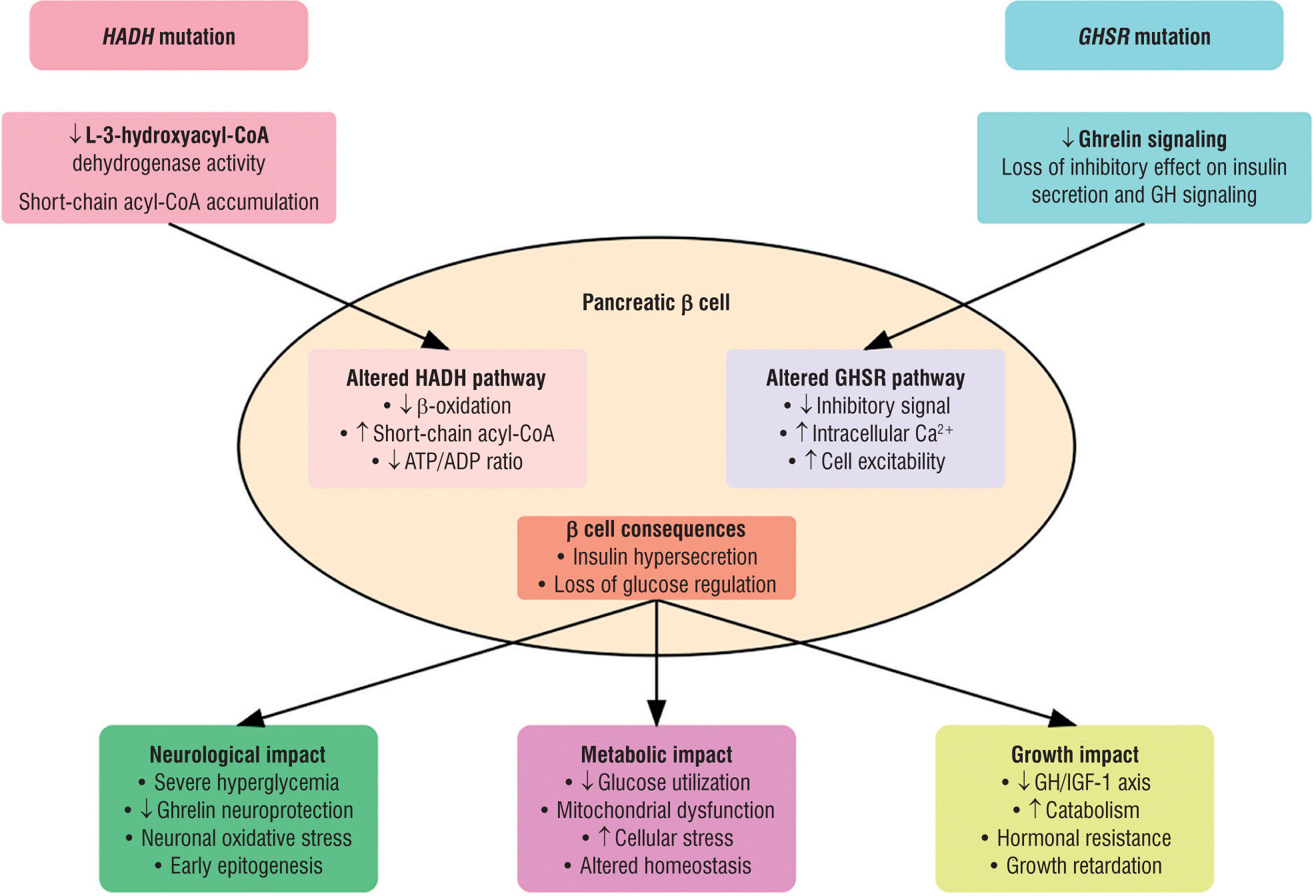
This unique form of congenital hyperinsulinism, associating mutations in HADH and GHSR genes, highlights complex and interconnected pathophysiological mechanisms. At the cellular level, there is a synergistic effect on insulin secretion, where HADH mutation disrupts fatty acid metabolism while GHSR mutation compromises ghrelin’s inhibitory signaling, together leading to amplified dysregulation of insulin secretion. The extensive metabolic abnormalities observed – including hyperaminoaciduria, elevated glycine, disturbed acylcarnitine profile, hyperammonemia, and hyperlactatemia – directly reflect this dual metabolic insult, providing biochemical evidence of the profound energy dysregulation resulting from the combination of these mutations. In particular, the elevated free carnitine and acetylcarnitine levels along with hyperglycemia likely reflect the HADH mutation’s impact on fatty acid oxidation, while the endocrine profile (low GH with low IGF-1) aligns with GHSR deficiency. The impact on neurological development is particularly marked, combining the loss of ghrelin’s neuroprotective role with increased vulnerability to hypoglycemic episodes and chronic cellular energy disruption. The consequences for growth are also significant, with altered GH/IGF-1 signaling via GHSR, a catabolic impact of chronic hyperinsulinism, and permanent cellular metabolic stress [3, 4, 9]. The proposed pathophysiological mechanisms underlying the severe phenotype in this HADH-GHSR digenic form are illustrated in Fig. 1.
Figure 1
Pathophysiological mechanisms of HADH-GHSR mutations in congenital hyperinsulinism. The diagram illustrates the cellular consequences of combined HADH and GHSR mutations in pancreatic β cells. HADH mutation leads to decreased L-3-hydroxyacyl-CoA dehydrogenase activity and accumulation of short-chain acyl-CoA, while GHSR mutation results in impaired ghrelin signaling. These alterations converge to cause insulin hypersecretion through distinct but complementary pathways, leading to neurological, metabolic, and growth impacts. Arrows indicate increase (↑) or decrease (↓) in pathway activity
ADP – adenosine diphosphate; ATP – adenosine triphosphate; GH – growth hormone; IGF-1 – insulin-like growth factor 1
This discovery of a digenic form opens important perspectives in several areas. It allows better understanding of complex interactions between fatty acid metabolism and hormonal signaling, suggesting the existence of previously unexplored metabolic regulation pathways. Moreover, it identifies potential new therapeutic targets that could improve the management of patients with severe forms of congenital hyperinsulinism. Finally, this observation underlines the crucial importance of extended genetic screening in atypical or severe forms of the disease, allowing better characterization of pathogenic mechanisms and more adapted management.
Methodological limitations include the pending completion of parental genetic testing, which would elucidate inheritance patterns of the HADH and GHSR variants. Additionally, the homozygous HADH deletion, while detected through exome-based CNV analysis, awaits confirmation by orthogonal methods due to technical constraints. These investigations are planned as part of ongoing genetic characterization of this novel digenic form.
The multisystemic complications observed (orthopedic, digestive, respiratory) emphasize the necessity of early multidisciplinary management and close monitoring of patients presenting with complex genetic forms of congenital hyperinsulinism.









