
Introduction
Primary biliary cholangitis (PBC) is a chronic autoimmune cholestatic liver disease involving the small intrahepatic bile ducts, leading to chronic cholestasis and its complications [1]. It most frequently affects women between 40 and 70 years of age [2]. PBC may be asymptomatic or present with pruritus, jaundice, and persistent fatigue [3, 4]. In advanced stages, steatorrhea and malabsorption, particularly of fat-soluble vitamins, are observed [5-7]. The etiology of primary biliary cholangitis remains unknown; however, genetic susceptibility and environmental factors are considered, including cigarette smoking, hormone replacement therapy, infections (especially urinary tract infections), exposure to xenobiotics, use of nail polish or hair dyes, and deficiencies in vitamin D and selenium [8, 9]. Characteristic immunological markers include antimitochondrial antibodies type 2 (AMA-M2) and, less frequently, antinuclear antibodies sp100 and gp210 [8-11]. The progressive inflammatory response destroys the small bile ducts and impairs bile outflow. The increasing accumulation of bile acids within hepatocytes further exacerbates inflammation, resulting in progressive cholestasis and ductopenia [12, 13]. If untreated, PBC can progress to cirrhosis with portal hypertension and hepatic failure and may ultimately be fatal [8, 9, 14]. Early diagnosis is crucial because timely therapy can prevent disease progression [15]. Ursodeoxycholic acid (UDCA) is the first-line treatment and, in most patients, delays disease progression [16, 17].
The aim of this paper is to review osteoporosis as a major extrahepatic complication of PBC by summarizing its prevalence and fracture burden, explaining the key disease-specific mechanisms linking cholestasis to impaired bone formation, and outlining practical approaches to risk assessment, screening, and treatment to prevent fragility fractures in patients with PBC.
Osteoporosis
Osteoporosis is a systemic skeletal disease characterized by increased bone fragility, decreased bone mineral density (BMD), and deterioration of bone microarchitecture, resulting in an elevated fracture risk and potentially reduced life expectancy. Low-energy fractures are typical and most commonly involve the femoral neck, forearm, and vertebrae, impairing function and markedly reducing quality of life [18, 19]. Osteoporosis occurs most commonly in postmenopausal women, but it also affects younger women and men [18]. Although men have a lower overall fracture incidence, they experience higher post-fracture mortality and complication rates [18, 20].
Risk factors for osteoporosis differ between women and men and are influenced by genetics, hormonal status, age, diet, physical activity, sleep hygiene, and socioeconomic status (Table 1) [21, 22]. The relationship between body mass index (BMI) and BMD remains debated; however, higher BMI is generally considered protective [22]. In premenopausal women, secondary causes are relatively common and include glucocorticoid use, anorexia, celiac disease, hyperthyroidism, hypogonadism, chronic kidney disease, diabetes mellitus, rheumatoid arthritis, anticonvulsant therapy, and malabsorption syndromes [23, 24]. Postmenopausal osteoporosis, the most prevalent form, is caused primarily by estrogen deficiency.
Table 1
Osteoporosis in primary biliary cholangitis from cholestasis to fracture prevention
Osteoporosis develops from an imbalance between bone resorption and formation, leading to reduced bone mass [25]. The receptor activator of nuclear factor κB ligand (RANKL) plays a central role by binding the RANK receptor on osteoclast precursors, thereby promoting their differentiation, activation, and survival and increasing bone resorption [26].
Diagnosis relies on densitometric assessment of BMD, most commonly by dual-energy X-ray absorptiometry (DEXA) at the proximal femur, lumbar spine, and forearm. Osteoporosis is diagnosed when the T-score is −2.5 or lower [27, 28]. Fracture risk estimation complements BMD measurement and typically uses tools such as the FRAX calculator to estimate 10-year fracture probability based on BMD, clinical risk factors, and lifestyle variables [29].
Early identification of patients at high fracture risk is essential to initiate anabolic therapy when indicated [29]. Vitamin D and calcium supplementation are necessary but not sufficient as the sole therapy in older adults [30]. Most pharmacologic options are antiresorptive agents that inhibit osteoclast activity. First-line therapy for patients with osteoporosis and high fracture risk consists of bisphosphonates. Monoclonal antibody therapy targeting RANKL is an alternative, particularly effective for postmenopausal women with contraindications to other agents [31]. In severe osteoporosis, defined by two or more vertebral fractures or very low BMD, teriparatide is recommended and remains the only osteoanabolic medication currently available [31, 32].
Primary biliary cholangitis and osteoporosis
There is a correlation between primary biliary cholangitis and osteoporosis, although the underlying mechanisms remain incompletely understood; the evidence base on this association continues to grow [33]. PBC frequently occurs in patients with systemic rheumatic diseases such as Sjögren syndrome or rheumatoid arthritis [6, 34]. Autoimmune liver diseases coexist with rheumatic diseases in approximately 30% of cases [35]. Osteoporosis is among the most common complications of PBC, and its prevalence increases with disease progression [36, 37]. Reported rates exceed 20% among patients with PBC and reach up to 44% in those with cirrhosis listed for liver transplantation [7, 38, 39]. The risk of osteoporosis also increases after liver transplantation, with bone loss of 8-18% observed within 3-6 months following the procedure [7].
Consistent with these findings, studies in PBC cohorts show a higher fracture incidence than in general-population age- and sex-matched controls. The prevalence of fractures is approximately 10-20%, rising to about 22% among individuals awaiting liver transplantation [40, 41]. In one study, the most common fracture sites were the vertebrae and hip. Fractures at these locations impair daily functioning and may lead to complications such as deep vein thrombosis, pressure ulcers, and pneumonia [42]. In patients with PBC, post-fracture mortality is higher than in individuals without PBC, likely reflecting the burden of chronic liver disease and comorbidities [42].
Major risk factors for osteoporosis in PBC include older age, corticosteroid use, cirrhosis, and histological disease severity; in women, postmenopausal status is an additional risk factor (Table 1). Although systemic glucocorticoids are not standard therapy for isolated PBC, they are commonly used in patients with coexisting autoimmune diseases (e.g., rheumatic conditions), overlap syndromes, and in some post-transplant regimens. Data specifically related to the subgroup of “PBC + another autoimmune disease treated with steroids” are limited. However, several PBC cohort studies have evaluated steroid exposure as a risk factor for osteoporosis [41, 42]. Glucocorticoid therapy increases osteoporosis risk through well-established mechanisms, including: reduced bone formation, enhanced bone resorption, disrupted calcium balance, and increased fall risk due to steroid-related myopathy [20-25].
Disease stage exerts the greatest influence on bone loss: patients with more advanced PBC are substantially more likely to develop osteoporosis [3, 41]. Among those with stage 3 or 4 disease, morbidity increases up to fivefold compared with earlier stages, and the rate of bone loss accelerates with disease progression [41].
Cholestatic liver disease is a recognized risk factor for bone loss in cirrhosis. Additional contributors include malabsorption, hypogonadism, vitamin D deficiency, low physical activity, gut dysbiosis, and corticosteroid use [43, 44]. In the study by Wu et al., the causal relationship between osteoporosis and PBC was found to be independent of BMI, calcium levels, triglycerides, and selected sex hormones [45]. In contrast, Chen et al. found that osteoporosis in PBC was associated with elevated bilirubin and prothrombin time (PT), decreased albumin, and features of portal hypertension – splenomegaly, esophageal varices, thrombocytopenia, and ascites – as well as fatigue [3]. A higher C-reactive protein-to-albumin ratio (CAR) is also associated with an increased risk of osteoporosis and may serve as a predictive biomarker in PBC [46].
Genetic susceptibility may contribute to both chronic liver disease and musculoskeletal disorders. The heritability of PBC is estimated at approximately 37.2%, and that of osteoporosis at around 25.9% [47]. Genetic factors linked to reduced bone mass include variants in the vitamin D receptor (VDR), the type I collagen α1 chain gene (COL1A1), and microsatellite repeat polymorphisms in the insulin-like growth factor-1 (IGF-1) gene, although their overall effect appears modest [41, 48]. Vitamin D levels themselves do not seem to meaningfully affect bone fragility in chronic liver disease, whereas VDR polymorphisms are associated with osteoporosis in PBC and independently predict reduced bone mineral density [41, 48, 49].
Insulin-like growth factor-1, synthesized in the liver, plays a key role in longitudinal bone growth and skeletal maturation and positively influences bone mass accrual [50]. In advanced PBC, reduced synthesis of IGF-1 – a circulating osteoblast-stimulatory factor, contributes to osteoblast dysfunction [41]. In the study by Saeki et al., lower serum IGF-1 levels were associated with a higher prevalence of osteoporosis and increased fracture risk in PBC [51]. The Sp1 polymorphism of COL1A1 also correlates with lower baseline bone mineral density and is recognized as a genetic marker of peak bone mass in PBC [41, 52, 53]. Variants of the claudin-14 (CLDN-14) gene have similarly been linked to reduced bone mineral density in PBC [41, 54].
Elevated bilirubin and bile acid levels observed in PBC negatively affect osteoblasts and may lead to their dysfunction. Serum from jaundiced individuals has a demonstrably detrimental effect on osteoblasts [55]. Bilirubin and lithocholic acid (LCA) reduce osteoblast viability, inhibit differentiation and mineralization, and induce apoptosis [56, 57]. UDCA, used in the treatment of PBC, neutralizes the harmful effects of bilirubin and LCA, stimulates osteoblast differentiation and mineralization, prevents osteoblast apoptosis, and increases osteoblast survival [55-58].
According to available data, both PBC and osteoporosis may be associated with gut microbiota dysbiosis, which is implicated in various intestinal and extraintestinal diseases, including autoimmune disorders [37, 59, 60]. The relationship between the microbiota and PBC pathogenesis and treatment is not yet fully defined [61]. However, emerging evidence links microbiota alterations to bone mineral density and to the course of postmenopausal osteoporosis, particularly through regulation of Th17 and Treg lymphocytes that are central to bone metabolism [62, 63]. The gut microbiota may also contribute to bone loss in chronic liver diseases, potentially via short-chain fatty acids (SCFAs) [64]. Butyrate and propionate inhibit osteoclastogenesis and prevent bone mass loss. They likely also stimulate osteoblast activity and modulate T-cell function [65].
Pathogenesis of osteoporosis in PBC
The pathogenesis of osteoporosis in PBC is complex and remains incompletely understood [66]. Importantly, the underlying pathophysiology differs from that of postmenopausal osteoporosis, in which increased bone resorption predominates. In PBC, osteoporosis is caused primarily by reduced bone formation. Nonetheless, exceptions exist – among postmenopausal women and individuals with hypogonadism, increased bone resorption may predominate [3, 38, 41].
A central role is played by the triad of osteoprotegerin (OPG), RANKL, and the RANK receptor (Fig. 1) [37, 40]. In PBC, low RANKL and elevated OPG levels have been reported [41]. OPG is osteoprotective, whereas RANKL induces osteoclastogenesis and contributes to bone loss; however, low RANKL may also reflect diminished osteoblast activity and low bone turnover, which increases bone fragility and fracture risk [37, 41]. Elevated OPG can be interpreted as a compensatory response to bone loss [41]. OPG inhibits RANKL-mediated osteoclast activation [37]. Some studies also suggest that, with PBC progression, hepatic OPG secretion may be inhibited, potentially increasing osteoclast activity and bone resorption [40]. Patients with PBC exhibit elevated levels of the inflammatory cytokines IL-1, IL-6, and TNF-α, which modulate the RANKL-RANK-OPG system and thereby indirectly or directly promote osteoclast activation [37, 40]. Nonetheless, reduced bone formation appears to be the predominant mechanism [3, 40, 41].
Fig. 1
Pathogenesis of osteoporosis in primary biliary cholangitis (original figure; created by the authors). Elevated tumor necrosis factor α (TNF-α) promotes osteoclastogenesis and activates mature osteoclasts by acting on osteoclast precursors. Receptor activator of nuclear factor κB ligand (RANKL) binds its receptor RANK on osteoclast precursors and stimulates their differentiation into osteoclasts. Osteoprotegerin (OPG) blocks RANKL and the RANKL–RANK interaction, thereby inhibiting osteoclastogenesis. High concentrations of bilirubin and bile acids act directly on osteoblasts, inducing apoptosis and reducing differentiation, mineralization, and viability. Bile acids also act via the vitamin D receptor (VDR), preventing osteoblast differentiation and leading to dysfunction. Levels of insulinlike growth factor 1 (IGF-1) decline following loss of growth hormone (GH) receptors on hepatocytes, which directly impacts osteoblasts. The net effect is impaired osteoblast function, increased bone fragility, and reduced bone mass
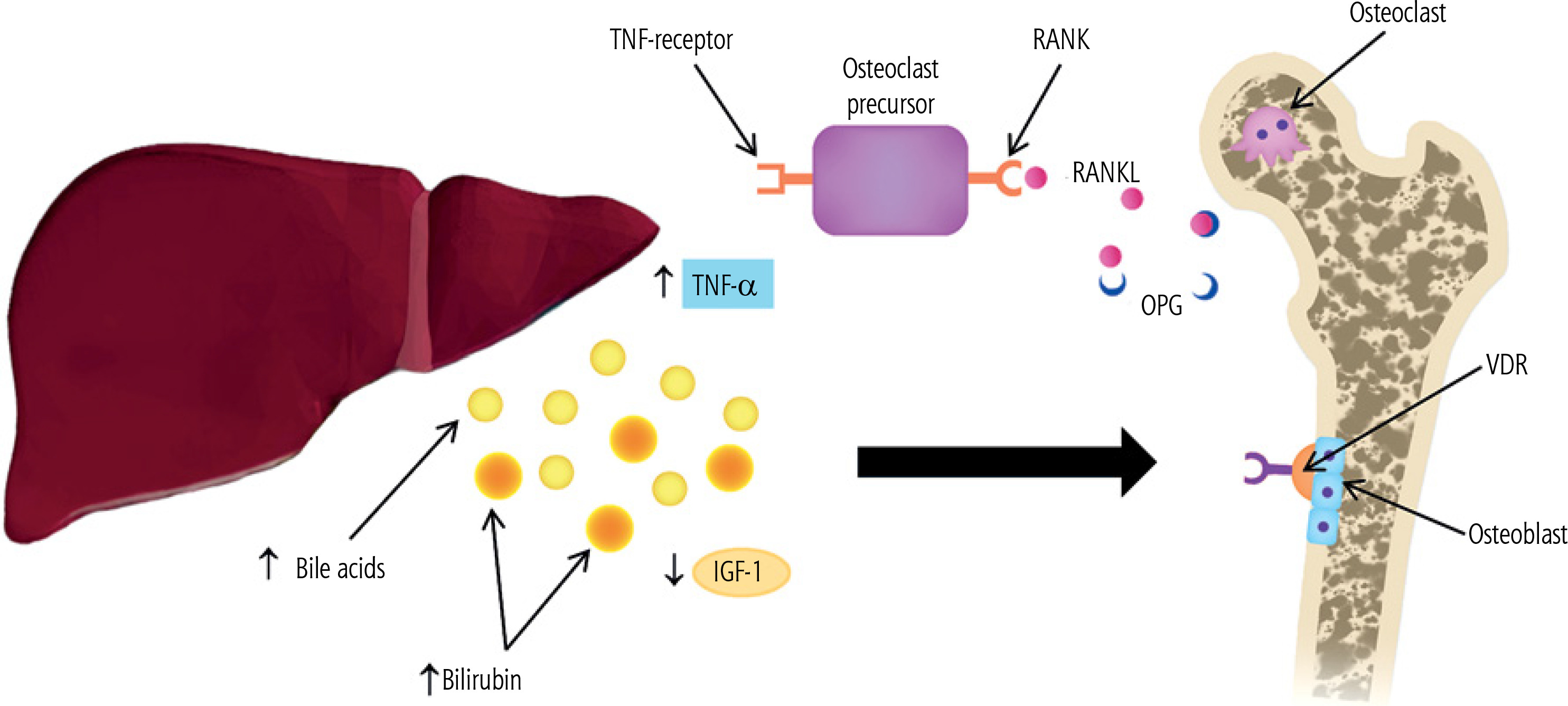
Bone resorption and formation are opposing processes, and disruption of this balance impairs bone mass accrual. When resorption exceeds formation, bone mass declines, leading to osteoporosis. Histomorphometric and microarchitectural studies in PBC indicate that osteoblast dysfunction and reduced bone formation are primary [3, 41]. Most patients with PBC and osteoporosis show reduced bone formation rates, decreased double tetracycline labeling (used to assess the rate of new bone formation and mineralization in osteoporosis diagnostics), fewer osteoblasts, and low serum osteocalcin levels [3, 38, 41]. Osteoblast dysfunction is associated with elevated bilirubin and bile acids, or may also result from the effects of alcohol on osteoblasts [3, 41, 67].
Lithocholic acid exerts both direct and indirect negative effects on osteoblast activity [41]. LCA interacts with the vitamin D receptor and modifies VDR-mediated gene expression, including regulation of RANKL, thereby contributing to osteoblast dysfunction [41, 68]. Impaired fibronectin production may likewise reduce bone formation by inhibiting osteoblast function in PBC [3, 38].
Osteoblast dysfunction may also reflect decreased circulating osteoblast-stimulatory factors such as IGF-1 [37, 41]. IGF-1 is synthesized primarily in the liver in response to growth hormone (GH) [37, 41, 50]. In chronic liver disease, progressive loss of GH receptors on hepatocytes leads to hepatic GH resistance and reduced serum IGF-1, which impairs osteoblast function and increases bone fragility [37, 41].
Primary biliary cholangitis carries a risk of malabsorption and deficiencies of the fat-soluble vitamins A, D, E, and K, which are important for coagulation and bone metabolism [40, 44]. Bile duct injury causes cholestasis, disrupting fat digestion and reducing absorption of these vitamins [42]. Vitamin A influences cellular growth and differentiation, including bone development; however, excess vitamin A adversely affects bone tissue by increasing osteoclast production, thereby reducing bone mass and elevating fracture risk [69, 70]. Vitamin D facilitates calcium absorption and bone mineralization, and its deficiency disrupts calcium homeostasis [44]. Vitamin E supports osteogenic differentiation, helps inhibit osteoclastogenesis, and protects bone cells from oxidative stress, which contributes to bone loss [44, 71]. Vitamin K functions as a coenzyme that activates non-collagenous bone proteins – osteocalcin (OC) and matrix Gla protein (MGP) – and promotes osteoblastogenesis while inhibiting osteoclastogenesis [38, 40]. In cholestasis and malabsorption, vitamin K deficiency leads to dysfunction of OC, MGP, and osteoblasts, thereby increasing the risk of osteoporosis [38, 40, 72].
Treatment
The evidence on the effectiveness of osteoporosis therapies specifically in PBC remains limited, and studies are scarce [73]. Given that reduced bone formation is the predominant pathogenic mechanism in PBC, antiresorptive drugs may be less effective. Consequently, current recommendations are based on data from postmenopausal osteoporosis [73].
According to European Association for the Study of the Liver (EASL) guidelines, lifestyle modifications are prioritized: an appropriate diet, avoidance of risk factors such as smoking, and engagement in weight-bearing/resistance exercise. Calcium and vitamin D supplementation may be considered; however, evidence for a clear benefit is lacking. Calcium should not be supplemented in individuals with adequate nutritional intake and no malabsorption, or in those with nephrolithiasis. Although no single, universally accepted threshold for treatment initiation is established, the EASL recommends therapy for patients with a femoral T-score below −1.5. Fracture risk should be assessed prior to treatment, for example using the FRAX calculator [13]. The EASL also states that bisphosphonates increase bone mass in PBC, particularly weekly alendronate and monthly ibandronate [74]. Caution is warranted with oral nitrogen-containing bisphosphonates because they can irritate the gastric or esophageal mucosa; special care is advised in patients with esophageal varices. Hormone replacement therapy (HRT) as menopause therapy is effective in postmenopausal women with PBC. HRT improves the vertebral bone density [75, 76]. Bone mineral density should be assessed at symptom onset and re-evaluated after 1-5 years, depending on DEXA results and overall fracture risk. The EASL further recommends assessing osteoporosis risk in all patients with PBC [13].
The American Association for the Study of Liver Diseases (AASLD) recommends BMD assessment every two years. Women should supplement calcium and vitamin D regardless of menopausal status – calcium 1500 mg/day and vitamin D 1000 IU/day – provided there is no history of nephrolithiasis. In advanced disease, serum vitamin D should be measured annually [11]. AASLD also supports the use of alendronate in patients with osteoporosis, with improvements in BMD similar to those observed with ibandronate [74]. Oral bisphosphonates should be avoided in patients with gastroesophageal reflux disease or esophageal varices. Although HRT improves BMD, AASLD notes that it is rarely recommended due to safety concerns [11, 76].
Ursodeoxycholic acid is first-line therapy for PBC [77]. UDCA improves survival and mitigates the deleterious effects of bilirubin and bile acids by promoting osteoblast differentiation and mineralization [42, 56, 58]. It enhances osteoblast activity and viability and prevents apoptosis [42, 55, 57]. However, UDCA has not been shown to reduce the incidence of osteoporotic fractures [42].
Second-line therapy in PBC includes fibrates, which act as peroxisome proliferator-activated receptor-α (PPAR-α) agonists [16, 78]. In combination with UDCA, fibrates improve liver biochemistry in PBC; depending on the agent, approximately 20-30% of treated patients achieve normalization of alkaline phosphatase (ALP), γ-glutamyltransferase (GGT), and total bilirubin [16, 17]. Fibrates also have a beneficial effect on bile acid metabolism and reduce serum bile acid levels [16, 79]. Data on skeletal effects are limited: fenofibrate has been shown to stimulate osteoblast differentiation via bone morphogenetic protein-2 (BMP2) expression [78, 80], but the available evidence is insufficient to establish a clear benefit for osteoporosis in PBC [78].
New PPAR-δ agonists, seladelpar and elafibranor, have demonstrated reductions in liver enzyme levels [81]. No data are currently available regarding their effects on osteoporosis.
An alternative option for the treatment of osteoporosis in patients with primary biliary cholangitis is denosumab. It is an effective and safe therapeutic option and, compared with zoledronic acid, is associated with fewer adverse effects [82].
Conclusions
Individuals with PBC have a substantially higher risk of fractures and an increased post-fracture mortality rate compared with general-population controls. Low energy fractures, particularly of the vertebrae and hip, are more frequent, adversely affecting both life expectancy and quality of life. Accordingly, fracture prevention strategies and education about skeletal risk are essential. Early diagnosis is critical, as it enables prompt treatment that may prevent disease progression and complications. Management of osteoporosis in PBC primarily focuses on bisphosphonates, alongside recommended vitamin D and calcium supplementation. UDCA, the standard therapy for PBC, may help slow bone loss by improving cholestasis and supporting osteoblast function.





