
Introduction
Hepatitis C virus (HCV) infection is one of the main causes of chronic liver diseases worldwide, including cirrhosis and hepatocellular carcinoma (HCC), contributing to a deterioration in both quality and length of life. Studies in recent years have shown that HCV infection also leads to the development of metabolic disorders, which play a significant role as a risk factor in the development of cardiovascular diseases. The notable impact of HCV on the development of obesity, insulin resistance, diabetes, lipid disorders, and steatotic liver disease has prompted some authors to refer to metabolic disorders in the context of HCV infection as a “metabolic-viral syndrome”, with steatotic liver disease described as the organ-specific manifestation of the metabolic syndrome. The observed increased incidence of cardiovascular disease among patients with chronic HCV infection has contributed to HCV now being regarded as a new, non-classical risk factor for cardiovascular diseases, with the associated complications considered an extrahepatic manifestation of HCV infection.
Cardiovascular diseases, which include ischaemic heart disease (coronary artery disease, myocardial infarction), cerebrovascular diseases (ischaemic stroke, transient ischaemic attack – TIA), and peripheral vascular diseases (aortic aneurysms, peripheral artery disease), account for about 4.3 million deaths in Europe each year, representing nearly half of all deaths – 48% (54% in women and 43% in men). In Poland, they account for 46% of all annual deaths (51.1% in women and 40.9% in men). They are also the leading cause of disability, hospitalisation, and rising healthcare costs in most European countries, including Poland.
In Poland, the main risk factors for atherosclerosisbased cardiovascular disease include hypertension, lipid disorders, obesity, diabetes, and smoking (Table 1).
Table 1
Classical risk factors for cardiovascular diseases
New risk factors also include infectious agents such as influenza virus infection, human immunodeficiency virus (HIV) infection, and HCV infection.
The potential role of HCV as a risk factor in the development of cardiovascular diseases is complex. On the one hand, the infection directly induces a chronic inflammatory state, contributing to the development of atherosclerosis and endothelial dysfunction; on the other hand, HCV infection indirectly leads to the development of other key risk factors such as insulin resistance, diabetes, obesity, hypertriglyceridaemia, hypertension, or chronic kidney disease [1, 2]. These metabolic disturbances described in HCV infection resemble those observed in the so-called metabolic syndrome (obesity, hypertension, diabetes or abnormal fasting blood glucose or glucose intolerance, elevated serum triglycerides or reduced serum HDL cholesterol), which is currently considered the main contributor to the rising risk of cardiovascular disease [3].
Considering the impact of HCV infection on the development of metabolic disorders and the risk of cardiovascular disease, it appears necessary for all HCV-infected patients to undergo regular periodic assessments of glucose levels (fasting blood glucose, oral glucose tolerance test – OGTT), a lipid profile, as well as blood pressure measurement, body weight control, and waist circumference measurement. Any detected abnormalities should be treated in accordance with current recommendations.
In HCV-infected patients referred for direct-acting antiviral (DAA) therapy, the potential for drug interactions that may affect treatment efficacy, dosing, or safety must be taken into account. Hence, it is extremely important to evaluate the risk of such interactions each time using www.hep-druginteractions.org. If a risk of serious interactions is identified, the planned HCV treatment regimen should be adjusted; if that is not possible, the previously used drugs should be replaced with safe alternatives or their dosages modified.
This is particularly important in HCV-infected patients with extreme, high, or very high cardiovascular risk (Table 2) who receive chronic lipid-lowering therapy or are treated with anticoagulants [4–7].
Table 2
[i] LDL – low-density lipoprotein, HDL – high-density lipoprotein, HDL-C – HDL cholesterol, hs-CRP – high-sensitivity C-reactive protein, ACS – acute coronary syndrome, ASCVD – atherosclerotic cardiovascular disease, ACR – albumin-to-creatinine ratio, eGFR – estimated glomerular filtration rate. *Patients with type 1 diabetes over 40 years of age can be classified into the respective risk groups similarly to those with type 2 diabetes. ACR – albumin-to-creatinine ratio in a single morning urine sample, ASCVD – atherosclerotic cardiovascular disease, non-HDL-C – total cholesterol (TC) – HDL-C
HCV infection as a cardiovascular risk factor
The global spread of hepatitis C virus (HCV) infection, responsible for millions of chronic hepatitis cases worldwide, has led to numerous studies on both hepatic and extrahepatic consequences in various patient cohorts. Morbidity and mortality associated with the progressive destruction of liver parenchyma, the development of portal hypertension, and organ failure are well recognised. However, long-term observations of patients with HCV infection have provided extensive evidence that the infection causes numerous extrahepatic manifestations related to autoimmune reactions, stimulation of the immune system, the development of mixed cryoglobulinaemia, and the lymphotropism of HCV. In recent years, multiple reports have confirmed a relationship between infection and metabolic disorders, importantly, not only as a result of progressive liver dysfunction.
The occurrence of steatotic liver disease associated with HCV infection, especially genotype 3, is the most widely known example, but steatotic liver disease may also be triggered by insulin resistance or type 2 diabetes, which is a known extrahepatic manifestation of HCV infection. These metabolic disturbances arise because of the virus’s capacity to affect lipid metabolism and insulin signalling pathways. This activity of HCV has been demonstrated in experimental research, and indirect evidence is provided by the resolution of metabolic disorders following viral eradication. In recent years, increasing attention has been paid to the relationships between HCV infection and cardiovascular disease (CVD). Naturally, the relatively high prevalence of CVD risk factors in the general population complicates any objective assessment of the existing pathophysiological links between the virus and cardiovascular disease, both indirect (in connection with the aforementioned metabolic disturbances) and direct. The existence of these relationships is particularly important given the availability of highly effective DAA treatments. In such cases, virus eradication should be considered a strategy that can reduce the risk of CVD.
HCV can promote atherogenesis by several mechanisms, both direct and indirect. Atherosclerotic plaques develop and destabilise due to persistent inflammatory changes. Chronic HCV infection causes hepatic inflammation as well as systemic inflammation. There is considerable evidence of cytokine release and increased oxidative stress. On the other hand, the fact that HCV RNA sequences have been isolated from carotid atherosclerotic plaques supports the hypothesis that HCV has a direct pro-atherogenic effect by inducing arterial inflammation, probably via the pro-inflammatory cytokine interleukin 1β. HCV proteins, both structural and non-structural, play a role in initiating and maintaining chronic inflammation. In addition, HCV disrupts the Th1/Th2 balance, involving cell-mediated immunity promoted and maintained by interleukin (IL)-2, tumor necrosis factor α (TNF-α), and interferon γ, vs. humoral immunity supported by IL-4, IL-5, IL-6, and IL-10. Mixed cryoglobulinaemia, considered the most common extrahepatic manifestation of HCV infection and a cause of multiple organ and systemic pathologies (among others, vasculitis), is also of particular significance.
HCV infection and the risk of carotid artery atherosclerosis and stroke
As early as 2003, reports indicated an association between HCV infection and carotid artery atherosclerosis. In ultrasound examinations, HCV-infected patients had an increased intima-media thickness (IMT) of the carotid arteries and a higher prevalence of atherosclerotic plaques compared with control subjects (64% vs. 25%), even after adjusting for metabolic risk factors in both groups. Subsequently, many studies worldwide have confirmed a higher prevalence of carotid atherosclerosis in the course of HCV infection. Although numerous studies have shown a significant link between carotid artery atherosclerosis and HCV infection, some isolated reports question such an association. Meta-analyses clarify these doubts: HCV infection more than doubles the risk of carotid atherosclerosis, whether measured by the presence and number of plaques or by IMT. This underscores the importance of continuing lipid-lowering therapy in this patient group.
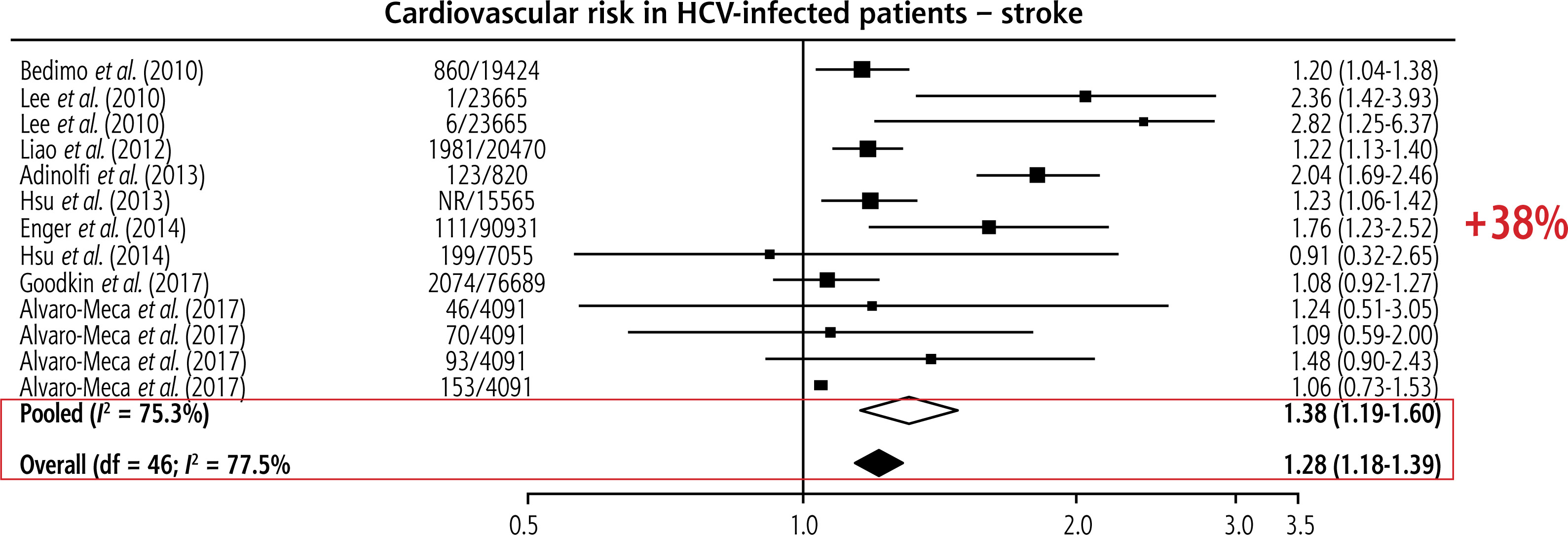
Most, though not all, studies confirm an association between HCV and stroke, especially ischaemic stroke. In a large retrospective population-based study, Enger et al. found an association between HCV infection and stroke (OR = 1.76, 95% CI: 1.23-2.52) [8]. Similarly, another retrospective study by Gutierrez et al. reported a strong association between HCV infection and stroke (OR = 9.61, 95% CI: 2.51-35.78) in NHANES cohort participants from 2005 to 2010 [9]. Overall, patients with HCV have a significantly greater risk of stroke than those without HCV (Fig. 1) [10].
HCV infection and the risk of coronary artery atherosclerosis
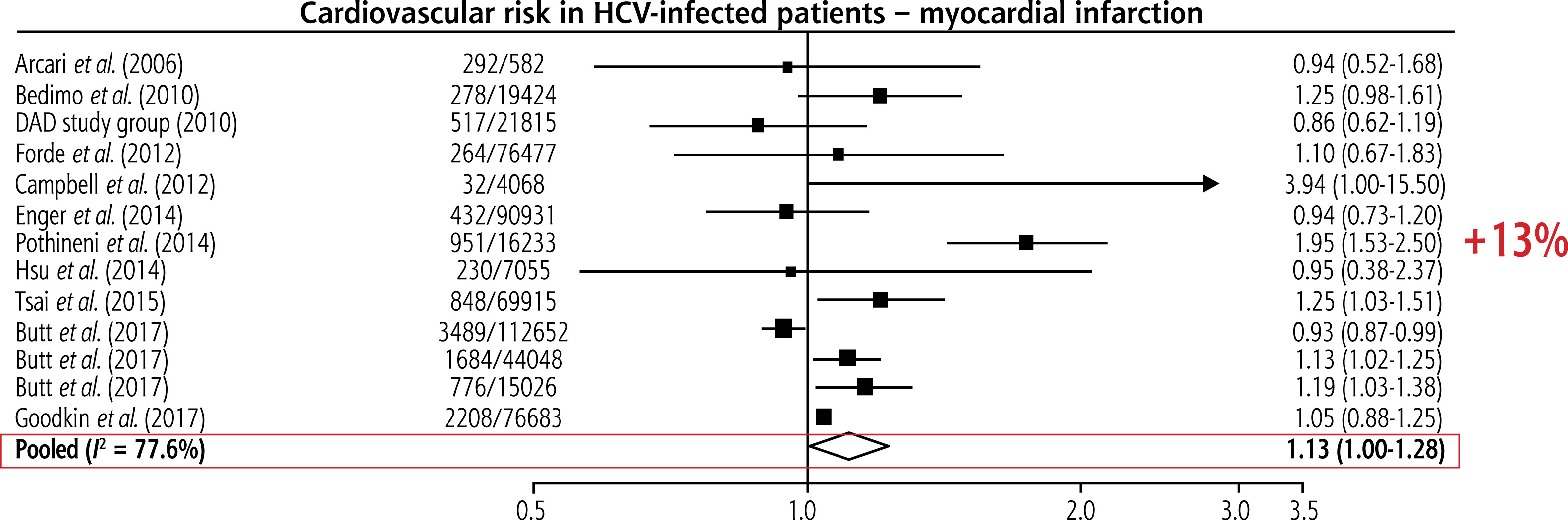
Also at the beginning of the 21st century, reports emerged regarding the risk of developing coronary artery disease (CAD) in individuals infected with HCV [10]. Research findings indicate that patients with HCV-related steatotic liver disease and dyslipidaemia, regardless of viral genotype, age, sex, and, importantly, the histological degree of liver damage, showed the highest incidence of atherosclerosis. Numerous studies suggest that HCV infection should be considered a risk factor for subclinical atherosclerosis. Overall, HCV patients have a 13% higher risk of myocardial infarction than people without HCV (Fig. 2).
HCV infection and the risk of arrhythmias
As early as 2004, Japanese researchers showed that HCV infection may be associated with ventricular enlargement, cardiac dysfunction, and myocardial fibrosis, leading to dilated cardiomyopathy, heart failure, and arrhythmias [11].
Besides cardiomyopathy, rhythm disorders in HCV-infected patients may be related to thyroid dysfunction, which is one of the extrahepatic manifestations of this infection. Moreover, HCV-infected individuals may have higher comorbidity rates compared to the general population. On the other hand, in a recently published large study conducted in Taiwan, after antiviral therapy, the incidence of new-onset atrial fibrillation (NOAF) decreased from 6% to 1.2% [12]. This interesting observation suggests that HCV treatment may be associated with a reduced risk of NOAF. In 2019, Wu et al., in an analysis covering 5,480 patients with HCV infection over a 13-year period, found that the risk of cardiomyopathy and arrhythmias was higher in HCV-infected individuals than in HBV-infected patients [13]. Regarding atrial and ventricular arrhythmias, the risk of sick sinus syndrome was significantly higher in patients with chronic HCV infection compared to those with chronic HBV infection. In addition, an increased frequency of cardiac pacing system implantation was observed in patients with chronic HCV infection, although it did not reach statistical significance.
Direct-acting antiviral drugs against HCV
Until recently, the treatment of chronic hepatitis C was based on injections of interferon α (natural, recombinant, pegylated) in combination with ribavirin. Such treatment, lasting 24-48 weeks, was typically associated with efficacy not exceeding 50%, numerous contraindications and adverse effects, and often the requirement of multiple re-treatments. Moreover, these drugs frequently impaired the overall functioning of patients. In view of the above, the introduction of DAA therapy against HCV was a real breakthrough for both patients and clinicians.
Although the first DAAs, belonging to the NS3/4A protease inhibitors (telaprevir and boceprevir), introduced in 2011, still required concurrent administration of interferon and ribavirin, they increased treatment efficacy, measured as a sustained virological response (SVR), to 65-75% [14]. The real revolution came four years later with the introduction of oral DAAs (so-called all oral therapies) that did not require interferon [15]. These therapies, adopted practically from the outset in Poland, almost completely eliminated adverse effects and offered treatment efficacy usually exceeding 95%, particularly in genotypes other than HCV GT3 [16]. DAA drugs are based on three classes that affect different elements of the viral replication cycle: protease inhibitors (NS3/4A), polymerase inhibitors (NS5B), and replication complex inhibitors (NS5B); see Table 3. Skilful combination of these drugs results in very rapid inhibition of viral replication. In the first Polish real-world Amber study, complete undetectability of HCV-RNA after the first week of treatment was observed in as many as around 25% of treated patients [17]. Rapid suppression of replication significantly reduces the risk of failure due to the emergence of DAA-resistant HCV variants, and the key determinant of treatment success is patient adherence.
Table 3
Currently recommended drugs for HCV infection therapy by class [18]
Another and final milestone in HCV treatment was the introduction of currently used pangenotypic regimens. Pangenotypic drugs show comparable efficacy in the treatment of HCV genotypes 1-6 (> 97% SVR12), making the treatment of traditionally so-called difficult-to-treat patient groups (including those infected with genotype 3 who have liver cirrhosis and a history of previous treatment failure) no longer a challenge. Currently, pangenotypic therapies account for over 98% of all treatments and are based primarily on two-drug oral DAA combinations, namely sofosbuvir with velpatasvir (SOF + VEL) administered for 12 weeks, or glecaprevir with pibrentasvir (GLE + PIB) for 8 weeks. In cases of DAA treatment failure, a triple therapy including voxilaprevir (SOF + VEL + VOX) may be used. The choice of treatment regimen depends mainly on the presence of contraindications and potential drug interactions. In simpler terms, for HCV-infected individuals with liver failure as assessed by the Child-Pugh scale (class B or C), GLE + PIB is not recommended, whereas in patients with eGFR < 30 ml/min/1.73 m2 or those on dialysis, SOF-based therapies are not recommended. At the same time, evaluating potential drug interactions is very important, because both SOF + VEL and GLE + PIB are substrates/inhibitors of drug transporters – P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and organic anion transporting polypeptide (OATP) – and are metabolised to varying degrees via different cytochrome P450 (CYP) isoforms. It is worth noting that, unlike other DAAs, SOF is largely (> 80%) excreted by the kidneys. A detailed description of how each DAA relates to drug transporters and their metabolism can be found in the Summaries of Product Characteristics (SPCs), which goes beyond the scope of this paper. Nevertheless, it is important to emphasise that an analysis of potential drug interactions before starting DAA therapy is crucial, because concurrent administration of DAAs with strong inducers of P-gp or CYP can reduce DAA levels, resulting in treatment failure. Conversely, the use of DAAs together with agents that inhibit P-gp, BCRP, OATP, or CYP can increase systemic DAA levels, leading to potential toxicity. There are also numerous drugs, such as amiodarone or digoxin, whose levels may rise dangerously when combined with some DAAs, causing serious adverse effects. Many of the lipid-lowering or anticoagulant agents discussed below also fall into this category.
Lipid-lowering therapy in patients receiving DAAs
The main goal in treating lipid disorders is the reduction of low-density lipoprotein cholesterol (LDL-C). It has been shown that lowering LDL-C by 1 mmol/l is associated with as much as a 20-24% reduction in the risk of cardiovascular events and overall mortality. The 2019 guidelines of the European Society of Cardiology and the European Atherosclerosis Society (ESC/EAS), subsequently updated by the 2021 guidelines of the Polish Society of Lipidology and five other scientific societies, strictly define target LDL-C levels (Table 2), which in high-, very high-, and extremely high-risk groups can only be achieved through intensive lipid-lowering treatment and/or (immediate) combination therapy with statins and ezetimibe (and possibly PCSK9 inhibitors), using atorvastatin or rosuvastatin at the highest permissible doses – 40-80 mg/day and 20-40 mg/day, respectively [19–21]. However, in some situations, even at such high doses, the cholesterol-lowering effect of 5-hydroxy-3-methylglutarylcoenzyme A (HMG-CoA) reductase inhibitors may be insufficient. In that case, ezetimibe should be added, followed by PCSK9 inhibitors (alirocumab, evolocumab) or the siRNA agent inclisiran [6, 7]. As noted above, especially in patients at very high cardiovascular risk, complex lipid-lowering therapy – primarily based on a statin and ezetimibe (ideally as a fixed-dose combination product) – can be considered immediately, without waiting for the effect of statins alone, in order to achieve the patient’s LDL-C target as soon as possible [5, 22].
Currently, simvastatin therapy should not be initiated in patients at extremely high, very high, or high risk, due to a weaker lipid-lowering effect compared to atorvastatin or rosuvastatin and because of the higher risk of simvastatin-associated adverse events (Tables 4 and 5) [23]. The lipid-lowering potential of pitavastatin at a dose of 4 mg is 43-47%, meaning that at least a 50% reduction can be achieved in around 30% of patients. Hence, the latest recommendations from the International Atherosclerosis Society recognise pitavastatin as a highest-intensity agent, while recommendations from a Polish expert group classify it as moderate- to high-intensity therapy [24, 25]. Total statin intolerance affects fewer than 10% of patients, and if recognised according to established definitions, it concerns about 5-7% [26]. The most common reason for statin intolerance is muscle-related symptoms, while elevated liver enzyme activity is less common and is transient in 95% of cases. For such patients, alternative therapy includes bempedoic acid (not widely available in Poland – only on a named patient basis), iPCSK9, or inclisiran in monotherapy or in combination with ezetimibe [6, 7, 20, 21].
Table 4
Comparison of equivalent statin doses in terms of lipid-lowering effect [23]
| Rosuvastatin | Atorvastatin | Pitavastatin | Simvastatin |
|---|---|---|---|
| – | – | – | 5 mg |
| – | – | – | 10 mg |
| 5 mg | 10 mg | 2 mg | 20 mg |
| 5-10 mg | 20 mg | 4 mg | 40 mg |
| 10-20 mg | 40 mg | – | – |
| 20 mg | 80 mg | – | – |
| 40 mg | – | – | – |
Table 5
Potential interactions of statins available in Poland with DAAs (based on https://www.hep-druginteractions.org/)
| Simvastatin | Atorvastatin | Rosuvastatin | Pitavastatin | |
|---|---|---|---|---|
| P-gp substrate | Yes | Yes | No | No |
| CYP3A4 substrate | Yes | Yes | No | No |
| OATP1B1 substrate | Yes | Yes | Yes | Yes |
| BCRP substrate | Yes | Yes | Yes | Yes |
| GLE/PIB | Not recommended | Not recommended | Potential risk of interaction Permitted maximum daily dose 5 mga Depending on the risk, consider combination therapy with ezetimibe and/or other non-statin drugs | Low risk of interaction Lowest dose 1 mg + monitoring Depending on the risk, consider combination therapy with ezetimibe and/or other non-statin drugs |
| SOF/VEL | Potential risk of interaction Switch statin to atorvastatin or rosuvastatin at permitted doses Depending on the risk, consider combination therapy with ezetimibe and/or other non-statin drugs | Potential risk of interaction Permitted maximum daily dose 40 mg Depending on the risk, consider combination therapy with ezetimibe and/or other non-statin drugs | Potential risk of interaction Permitted maximum daily dose 10 mg Depending on the risk, consider combination therapy with ezetimibe and/or other non-statin drugs | Low risk of interaction Monitor the patient and consider statin dose reduction or switching to atorvastatin or rosuvastatin at permitted maximum doses Depending on the risk, consider combination therapy with ezetimibe and/or other non-statin drugs |
| SOF/VEL/VOX | Not recommended Depending on cardiovascular risk, consider non-statin drugs | Potential risk of interaction Permitted maximum daily dose 20 mg Depending on the risk, consider combination therapy with ezetimibe and/or other non-statin drugs | Not recommended Depending on cardiovascular risk, consider non-statin drugs | Not recommended |
Among fibrates, fenofibrate is the safest (and essentially the only available in Poland) option for combination with a statin. This combination is used primarily in patients whose LDL-C target is already met but whose triglyceride (TG) level remains high (> 200 mg/dl, > 2.3 mmol/l). According to recommendations, it should be considered first in patients without coexisting cardiovascular disease, because of its strong potential to prevent both micro- and macrovascular complications. Icosapent ethyl (not available in Poland) or currently available omega-3 preparations at a dose of 2 × 2 g can be considered in patients at least at high risk, with persistent hypertriglyceridaemia of > 150-499 mg/dl (1.5-5.2 mmol/l) despite statin therapy and lifestyle modifications [20, 21].
An observed increased incidence of cardiovascular disease in patients with chronic hepatitis C has resulted in HCV being treated in recent years as a non-classical risk factor for cardiovascular diseases, with the associated complications viewed as an extrahepatic manifestation of HCV infection [27–29]. The potential role of HCV as a risk factor in the development of cardiovascular diseases is complex. On one hand, the infection directly leads to chronic inflammation, contributing to the development of atherosclerosis and endothelial dysfunction; on the other, it gives rise to metabolic disorders (obesity, insulin resistance, steatotic liver) that have long been recognised as classic cardiovascular risk factors.
The use of statins in patients with chronic hepatitis C is highly important. It has been shown that statins not only lower LDL-C levels but also have a strong anti-inflammatory effect. In preclinical and observational studies, they have been found to inhibit fibrotic processes in liver tissue, reduce portal pressure, and lower the risk of progression of liver disease and hepatocellular carcinoma, thereby improving survival in hepatitis C patients [30]. Moreover, their use does not elevate the risk of any adverse effects, including a clinically significant rise in liver enzyme activity. Hence, their use is strongly recommended in all patients with chronic liver disease, except in acute scenarios (for example, cirrhosis exacerbation) [20, 31, 32].
In HCV-infected patients referred for DAA therapy, treatment must account for potential drug interactions that may affect efficacy, dosage, or the safety of a given therapy. Therefore, it is extremely important to assess each time the risk of such interactions on the website www.hep-druginteractions.org. It should be emphasized that the risk of drug interactions does not necessarily mean that adverse effects caused by the concurrent use of the respective medications will occur. If there is a possibility of serious interactions, the planned HCV treatment regimen should be adjusted; if that is not feasible, the previously used drugs should be replaced with safer ones or their doses modified (Tables 5 and 6). The principle of balancing the benefit of treatment against the risk of discontinuation/dose modification for the patient must always be observed. This is particularly important in HCV-infected patients at extremely high, very high, or high cardiovascular risk for whom discontinuation of lipid-lowering therapy during DAA treatment is not an option.
Table 6
Potential interactions of other lipidlowering drugs with DAAs (based on https://www.hep-druginteractions.org/)
| Ezetimibe | Bempedoic acid | Fenofibrate | Alirocumab/Evolocumab | Inclisiran | |
|---|---|---|---|---|---|
| P-gp | Substrate | Not applicable | Not applicable | Not applicable | Not applicable |
| CYP3A4 | Not applicable | Not applicable | Not applicable | Not applicable | Not applicable |
| OATP1B1 | Substrate | Weak inhibitor of oatp1b1/oatp1b3 | Not applicable | Not applicable | Not applicable |
| BCRP | Not applicable | Not applicable | Not applicable | Not applicable | |
| GLE/PIB | Potential risk of interaction* | Potential risk of interaction** | No interaction | No interaction | No interaction |
| SOF/VEL | No interaction | No interaction | No interaction | No interaction | No interaction |
| SOF/VEL/VOX | Potential risk of interaction* | Potential risk of interaction** | No interaction | No interaction | No interaction |
P-gp – P-glycoprotein 1, CYP – cytochrome P450, OATP – organic anion transporting polypeptide, BCRP – breast cancer resistance protein, GLE – glecaprevir, PIB – pibrentasvir, SOF – sofosbuvir, VEL – velpatasvir, VOX – voxilaprevir
* Ezetimibe is primarily metabolised in the small intestine and liver by glucuronidation, and ezetimibe glucuronide is transported by the OATP1B1 protein. Increased exposure to this glucuronide can occur if OATP1B1 and P-gp inhibitors, such as GLE/PIB or VOX and VEL, are administered concurrently. In such cases, adverse effects should be monitored. According to the SPC, despite convincing clinical data, caution is advised when using ezetimibe in patients with moderate hepatic impairment
** Bempedoic acid and its glucuronide are weak inhibitors of OATP1B1 and OATP1B3. Concomitant administration of bempedoic acid and medicinal products that are substrates of OATP1B1 or OATP1B3 (for example, GLE, VOX) may lead to increased plasma concentrations of these medicinal products. If bempedoic acid and SOF/VEL/VOX must be used together, the patient should be monitored closely for adverse effects, particularly those related to elevated ALT levels. If bempedoic acid must be used, the recommended therapy for chronic hepatitis C should be SOF/VEL
Therapy with glecaprevir and pibrentasvir
Patients with chronic hepatitis C who qualify for treatment with GLE/PIB should not receive lipophilic statins – atorvastatin and simvastatin – due to the risk of serious drug interactions resulting from the inhibition of OATP1B1, P-gp, BCRP, and CYP3A by GLE and PIB, which increases serum levels of these statins. For atorvastatin, AUC increases 8.28-fold and Cmax increases 22-fold; for simvastatin, AUC increases 4.48-fold and Cmax increases 10.2-fold. This elevates the risk of myopathy, including rhabdomyolysis [33].
In the case of rosuvastatin therapy, AUC increases 2.15-fold and Cmax increases 5.62-fold due to inhibition of OATP1B1/3 and BCRP. The permitted maximum daily dose of rosuvastatin during GLE/PIB therapy in Europe is 5 mg/day. In this situation, in patients at high cardiovascular risk, combination therapy with nonstatin agents or change of antiviral therapy to SOF/VEL is recommended.
For pitavastatin, drug interactions have not been studied; however, because GLE/PIB inhibits OATP1B1, an increase in serum pitavastatin concentration is expected, and the dose should therefore be reduced. According to the SPC, therapy should be initiated at 1 mg/day with the possibility of increasing the dose. In patients treated with ezetimibe, potential adverse interactions may occur due to inhibition of OATP1B1 and P-gp by GLE/PIB. Adverse effects should be monitored [33].
Therapy with sofosbuvir and velpatasvir
Velpatasvir inhibits P-gp and BCRP, which, when combined with atorvastatin, increases its Cmax by 68% and AUC by 54%. The maximum permissible daily dose of atorvastatin in combination with SOF/VEL is 40 mg. In the case of rosuvastatin, due to inhibition of BCRP by VEL, rosuvastatin AUC increases by 170% and Cmax by 160%, so the permitted maximum daily dose of rosuvastatin in patients treated with SOF/VEL should not exceed 10 mg. For pitavastatin, drug interactions have not been studied; however, due to the inhibition of BCRP by SOF/VEL, an increase in its serum concentration is expected, and therefore the dose should be reduced. In this situation, in patients at high cardiovascular risk, combination therapy using permitted statins and non-statin agents is recommended. Patients treated with ezetimibe do not require a change in therapy if they qualify for SOF/VEL.
Therapy with sofosbuvir, velpatasvir and voxilaprevir
Rosuvastatin, simvastatin, and pitavastatin in combination with SOF/VEL/VOX are contraindicated.
For rosuvastatin, inhibition of BCRP by VOX causes increases in rosuvastatin serum concentrations (Cmax rises 18.9-fold, AUC rises 7.4-fold). Interactions with simvastatin and pitavastatin have not been studied; however, through the inhibitory effect of VOX on BCRP (in the case of simvastatin) and on OATP1B1 (in the case of pitavastatin), increases in serum statin concentrations, and with that the risk of myopathy, including rhabdomyolysis, can be expected.
SOF/VEL/VOX therapy, by inhibiting OATP1B1, increases atorvastatin concentrations (Cmax by 68%, AUC by 54%). As a result, the maximum permissible daily dose of atorvastatin is 20 mg. In this situation, for patients at high cardiovascular risk, combination therapy with atorvastatin and non-statin drugs is recommended. No studies have been conducted on interactions between ezetimibe and SOF/VEL/VOX. Because of the inhibitory effect on OATP1B1 and P-gp by VEL and VOX, an increase in serum ezetimibe concentration may be expected. Treatment is not contraindicated but should be monitored.
Summary of lipid-lowering therapy use in patients receiving DAAs
The safest DAA option for patients at extremely high, very high, and high cardiovascular risk is SOF/VEL, because it carries a low risk of interactions with lipid-lowering agents.
In patients at extremely high, very high, and high cardiovascular risk treated with SOF/VEL, the recommended statins are: atorvastatin at a maximum daily dose of 40 mg, rosuvastatin 10 mg, and pitavastatin with or without ezetimibe and other non-statin agents, so that the patient achieves the LDL cholesterol target.
In patients treated with SOF/VEL/VOX, the only recommended statin is atorvastatin at a daily dose not exceeding 20 mg.
Treatment with simvastatin or atorvastatin should be discontinued immediately if GLE/PIB therapy is planned, and consideration should be given to starting rosuvastatin at 5 mg or pitavastatin at an initial daily dose of 1 mg.
If bempedoic acid must be used, the recommended chronic hepatitis C therapy should be SOF/VEL.
If it is necessary to administer both bempedoic acid and SOF/VEL/VOX, it is advisable to monitor the patient for adverse effects, particularly those associated with increased ALT levels.
Regardless of the type of DAA, fenofibrate, iPCSK9, and inclisiran are safe lipid-lowering drugs.
Anticoagulant therapy in patients receiving DAAs
Non-vitamin K antagonist oral anticoagulants
Non-vitamin K antagonist oral anticoagulants (NOACs) are currently recommended as first-line anticoagulants [34, 35]:
in the treatment of venous thromboembolism (VTE), in the long-term prevention of VTE recurrence,
in the prevention of VTE after major orthopaedic surgery,
in the prevention of stroke in patients with non-valvular atrial fibrillation,
additionally, rivaroxaban 2 × 2.5 mg is indicated for the prevention of atherothrombotic events in patients following acute coronary syndrome (ACS), with stable coronary artery disease, or symptomatic peripheral artery disease (Table 7).
Table 7
Dosing of NOACs depending on the aim of treatment [34]
In non-valvular atrial fibrillation and VTE, continuation of dabigatran therapy is not recommended, particularly when using GLE/PIB or SOF/VEL/VOX.
GLE/PIB are strong P-gp inhibitors and therefore increase dabigatran Cmax (maximum concentration) 2.05-fold and its AUC (area under the curve) 2.38-fold [36]. Dabigatran exposure may also be raised by the mild P-gp inhibition exerted by VEL and VOX. Concomitant administration of dabigatran (75 mg as a single dose) with SOF/VEL/VOX (400/100/100 mg as a single dose) and VOX (100 mg as a single dose) increased dabigatran Cmax and AUC by factors of 2.87 and 2.61, respectively [37]. Dabigatran does not affect the efficacy of antiviral therapies (Table 8) [38].
Table 8
| Dabigatran | Rivaroxaban | Apixaban | |
|---|---|---|---|
| P-gp substrate | Yes | Yes | Yes |
| CYP3A4 substrate | No | Yes (moderate – 18%) | Yes (moderate – 20%) |
| OATP1B1 substrate | No | Yes | Yes |
| BCRP substrate | No | Yes | Yes |
| GLE/PIB | Not recommended Switch to apixabana | Potential risk of interaction Consider apixaban | Low potential risk of interaction Continue and monitor treatment |
| SOF/VEL | Potential risk of interaction Consider apixaban | Potential risk of interaction Consider apixaban | Low potential risk of interaction Continue and monitor treatment |
| SOF/VEL/VOX | Not recommended Switch to apixaban | Potential risk of interaction Consider apixaban | Low potential risk of interaction Continue and monitor treatment |
Rivaroxaban is a substrate of CYP3A4, P-gp, and BCRP [39]. Concomitant use of GLE/PIB can increase rivaroxaban concentrations through the additive inhibitory effect on CYP3A4, P-gp, and BCRP. In a large retrospective multicentre cohort study of patients receiving HCV DAAs and NOACs, including GLE/PIB and rivaroxaban, a low incidence of bleeding events was reported. Nonetheless, caution may be warranted when these agents are given together in patients with moderate or severe renal impairment. Patients should be reminded to promptly report any signs of bleeding or bruising, as is advised for all individuals prescribed NOACs. Rivaroxaban is contraindicated in patients with liver disease associated with coagulopathy and clinically relevant bleeding risk, including those with cirrhosis classified as Child-Pugh B or C [38].
Apixaban is primarily metabolized by CYP3A4/5 (with a minor role of CYP 1A2, 2C8, 2C9, 2C19, and 2J2) and is a substrate of P-gp and BCRP. Apixaban levels may rise owing to the strong inhibition of P-gp by GLE/PIB and inhibition of BCRP. However, in a large retrospective multicentre cohort study of patients who concurrently received HCV DAAs and direct oral anticoagulants (DOACs), including GLE/PIB and apixaban, the incidence of bleeding was low [38, 40].
In non-valvular atrial fibrillation, it is preferable to continue or switch the patient to apixaban therapy, coupled with more frequent blood counts and eGFR checks. It should be noted that apixaban is contraindicated in patients with liver failure classified as ChildPugh C (Table 9). In non-valvular atrial fibrillation with high bleeding risk, occlusion of the left atrial appendage should be considered prior to initiating DAA therapy, with a cardiology consultation required.
Table 9
Use of NOACs in patients with liver cirrhosis according to Child-Pugh class [34]
| Drug | Child-Pugh class | ||
|---|---|---|---|
| A score < 7 | B score 7-9 | C score > 9 | |
| Dabigatran | Standard dose Depending on platelet count and renal function | Caution is required | Not recommended |
| Apixaban | |||
| Rivaroxaban | Not recommended | ||
Vitamin K antagonists – warfarin, acenocoumarol
Warfarin and acenocoumarol (vitamin K antagonists) are absolutely indicated in the treatment and prevention of venous and arterial thrombosis in antiphospholipid syndrome and in the prevention of thromboembolic complications in patients with valvular atrial fibrillation (moderate or severe mitral stenosis) or with a mechanical heart valve.
Warfarin and acenocoumarol are metabolised by CYP2C9 and CYP1A2. GLE/PIB is a weak inhibitor of CYP1A2 and appears to be associated with a low risk of bleeding during vitamin K antagonist therapy (Table 10). It is important to remember that in patients with cirrhosis who are relatively compensated, liver function may improve while on DAA therapy, which is why more frequent INR checks are recommended (at least once a week). The most convenient option for the patient is home INR monitoring. Unfortunately, portable INR meters are not reimbursed in Poland.
Table 10
Potential interactions of warfarin and acenocoumarol with DAAs [41]
Table 11 presents the principles of switching between NOAC and heparin regimens.
Table 11
Antiplatelet agents
Regardless of which DAA is used, acetylsalicylic acid, prasugrel, and clopidogrel are considered safe antiplatelet agents (Table 12) [40, 42].
Table 12
Potential interactions of antiplatelet agents with DAAs
Ticagrelor is a substrate of CYP3A4 and P-gp, which can lead to potential interactions when combined with GLE/PIB, VEL, or VOX, thereby increasing ticagrelor levels and the risk of bleeding. A safer option is to switch from ticagrelor to prasugrel or clopidogrel (Table 13).
Summary of anticoagulant treatment in patients receiving DAAs
For patients with VTE treated with GLE/PIB, SOF/VEL, or SOF/VEL/VOX, the safest therapeutic option is low-molecular-weight heparin, unfractionated heparin, or fondaparinux (a synthetic, selective factor Xa inhibitor, which is not reimbursed).
Therapy with warfarin/acenocoumarol may be continued, but more frequent INR monitoring is recommended.
In patients with non-valvular atrial fibrillation, apixaban therapy should be considered regardless of the type of DAA.
Dabigatran therapy should be promptly discontinued if GLE/PIB or SOF/VEL/VOX are introduced, and apixaban should be started.
In patients with non-valvular atrial fibrillation and a high bleeding risk, left atrial appendage closure should be considered prior to commencing DAA therapy.
Regardless of the type of DAA, acetylsalicylic acid, prasugrel, and clopidogrel are safe antiplatelet agents.





