
Introduction
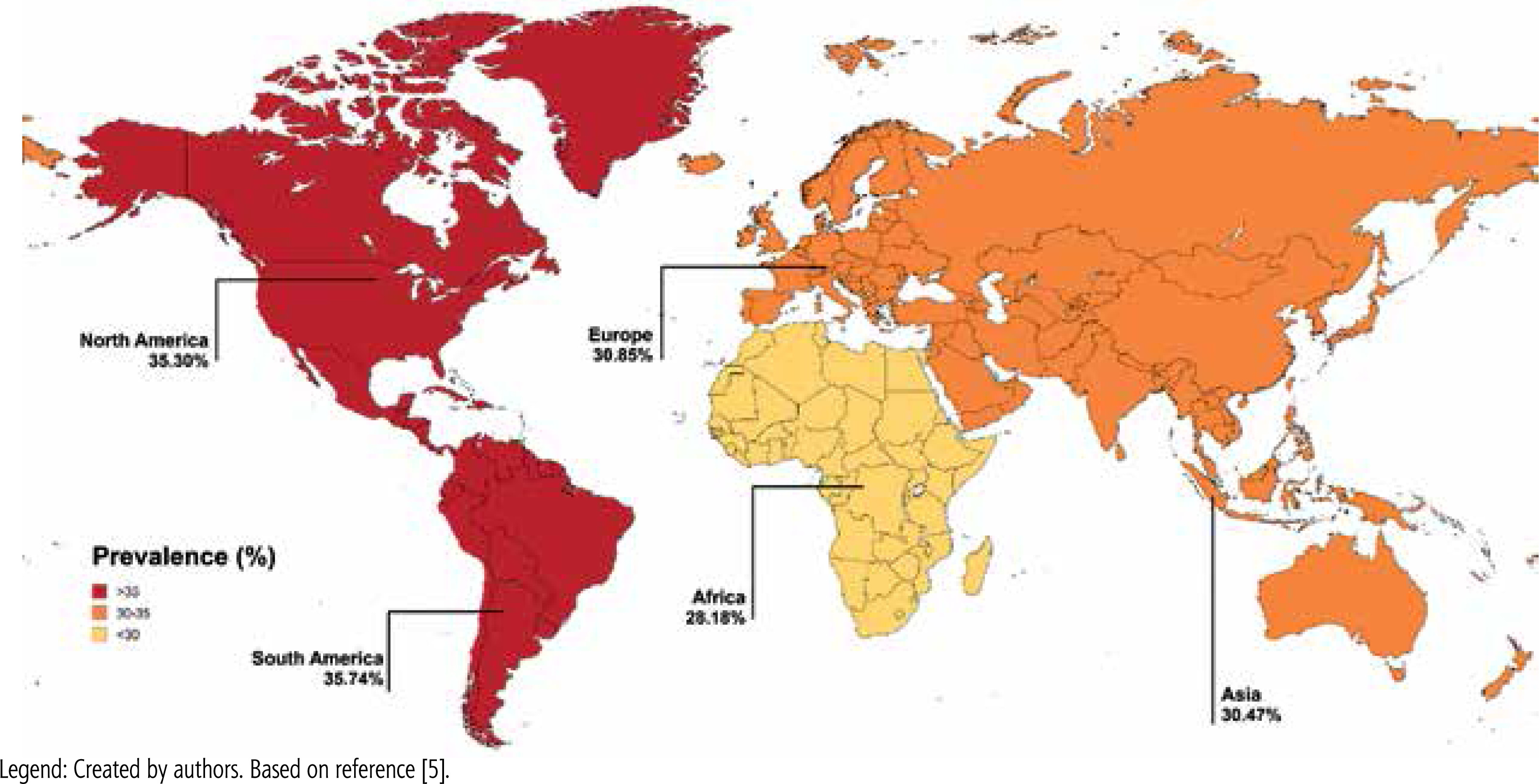
Steatotic liver disease (SLD) is an umbrella term for a wide range of conditions associated with metabolic dysfunction. SLD encompasses liver disorders characterized by excessive fat accumulation in hepatocytes, resulting from various causes such as metabolic disorders, monogenic diseases, drug-induced liver damage and alcohol abuse [1]. In recent years, there has been a change in the nomenclature and diagnostic framework for the disease, with the acronym SLD replacing the previously used term “nonalcoholic fatty liver disease” (NAFLD) in accordance with the 2023 Delphi Consensus [2]. The change, endorsed by hepatology experts, reflects a more comprehensive understanding of the metabolic basis of SLD and its strong association with systemic disorders, including obesity, type 2 diabetes mellitus (T2DM), dyslipidemia and hypertension [3]. The global burden of SLD has increased dramatically, fueled mainly by the obesity pandemic, making it the most common cause of chronic liver disease in recent years [4]. Currently SLD affects about 30% of the world’s population, more often men. Its distribution varies significantly across different regions of the world (Fig. 1) [4, 5]. While many cases of SLD remain clinically stable without significant progression, some may progress to severe forms of the disease, such as advanced liver fibrosis, cirrhosis with risk of liver failure, and hepatocellular carcinoma (HCC). The progressive form of SLD is defined as metabolic dysfunction associated steatohepatitis (MASH), which is characterized by liver inflammation and fibrosis resulting from the accumulation of fat in the liver [6]. The risk of progression becomes even higher when steatosis coexists with liver damage of other etiologies, including hepatitis B virus (HBV) and hepatitis C virus (HCV) infections [6]. Chronic viral hepatitis caused by HBV and HCV continues to be a significant contributor to liver disease and liver-related mortality.
Steatotic liver disease and chronic hepatitis C
Epidemiology
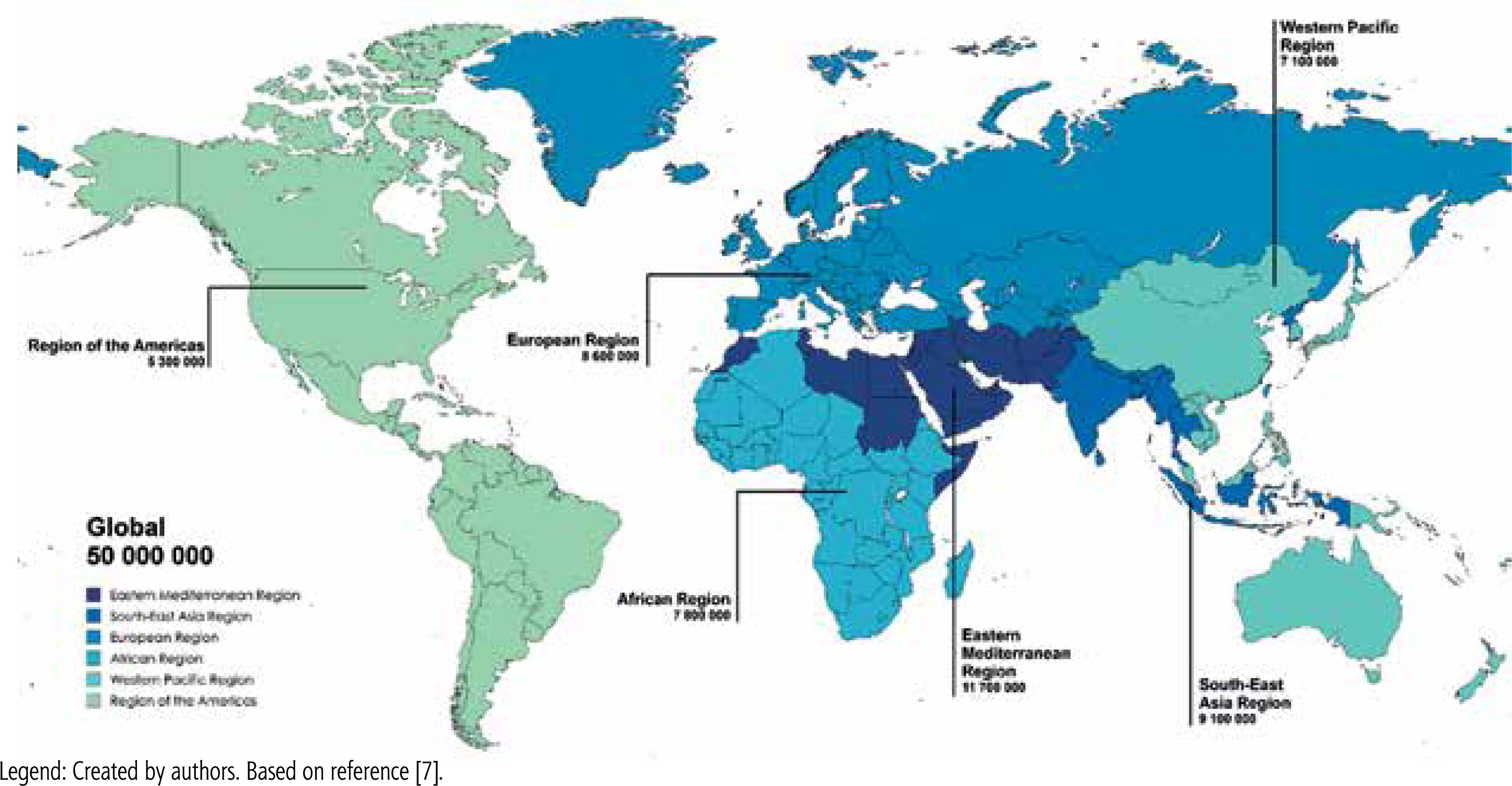
HCV infection remains a significant public health problem worldwide. According to the World Health Organization (WHO), about 50 million people are currently living with chronic HCV infection, making it one of the leading causes of chronic liver disease, cirrhosis and HCC (Fig. 2) [7]. Hepatic steatosis, defined as excessive lipid accumulation in hepatocytes, is a common histopathological feature in patients with chronic hepatitis C (CHC). Studies indicate that the prevalence of hepatic steatosis in patients with CHC ranges from 45% to 79%, which is significantly higher than in the general population [8]. Significant geographic differences have also been observed in the epidemiology of hepatic steatosis in patients with CHC. In countries such as France, Germany, Italy, and the United States, due to the increased prevalence of T2DM and obesity, steatosis associated with the metabolic syndrome predominate [8]. In South Asia and Australia, where there is a higher percentage of patients infected with HCV genotype 3, the predominant viral-related steatosis present in more than 50% of infected patients [9]. Ethnic and genetic factors may also influence the variation in the prevalence and severity of steatosis [8]. Although the most common form observed in patients with CHC is mild steatosis [10], its clinical consequences should not be underestimated. In HCV-infected individuals, steatosis is often associated with more advanced histological changes and faster progression of fibrosis [10]. Another emerging issue is steatohepatitis, in which, in addition to fat accumulation in liver cells, inflammation develops with damage to hepatocytes and their ballooning degeneration. It is estimated that steatohepatitis occurs in about 10% of patients with CHC [10].
Risk factors associated with the development of hepatic steatosis and progression of liver disease in patients with CHC
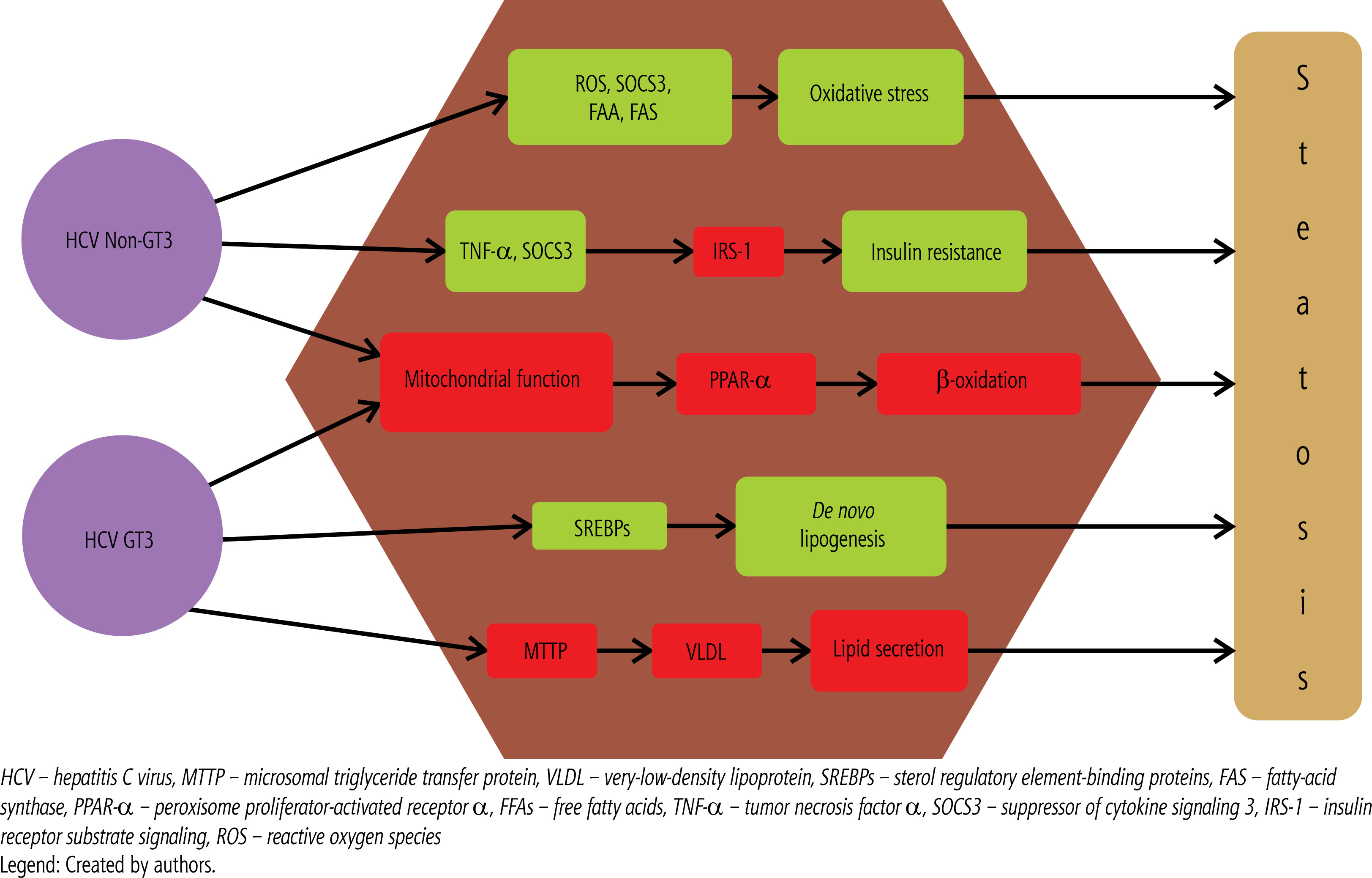
The development of hepatic steatosis in patients with CHC is determined by numerous risk factors, which can be broadly divided into viral and metabolic factors (Table 1). Steatosis associated with viral factors is mainly the result of direct effects of HCV, especially genotype 3, on lipid metabolism [11]. Figure 3 presents the main molecular mechanisms by which HCV induces steatosis, depending on the genotype. Steatosis associated with host metabolic disorders results from the influence of factors such as insulin resistance, obesity, and diabetes. However, these two forms of steatosis are not completely separate and often coexist, leading to complex interactions that make it difficult to determine their individual impact on liver disease progression [11].
Table 1
Differences between host- and virus-related steatosis in chronic hepatitis C
[i] CHC – chronic hepatitis C, GT3 – genotype 3, HCV – hepatitis C virus, MTP – microsomal triglyceride transfer protein, PPAR-α – peroxisome proliferator-activated receptor α, PTEN – phosphatase and tensin homolog, SLD – steatotic liver disease, SREBP-1 – sterol regulatory element-binding protein-1, T2DM – type 2 diabetes mellitus, VLDL – very low-density lipoprotein
Steatosis in patients with CHC associated with metabolic factors
Metabolic factors play a key role in the pathogenesis of hepatic steatosis in HCV, one of which is insulin resistance. Insulin resistance is considered a direct consequence of HCV infection and is the mechanism leading to lipid accumulation in the liver [11]. It results in increased hepatic glucose production, reduced lipid oxidation, and increased fat accumulation in hepatocytes [11]. A study of 500 patients with CHC found that insulin resistance (OR: 1.80, 1.15-2.81, p = 0.009) was a factor that independently increased the risk of developing advanced liver fibrosis [12]. One of the most significant metabolic risk factors for steatosis in CHC is T2DM. A meta-analysis of 34 epidemiological studies showed that HCV infection increases the risk of T2DM by about 68% compared to uninfected individuals [13]. This is likely related to changes in insulin signaling pathways, the development of insulin resistance, and chronic inflammation caused by the virus. Insulin resistance, hyperglycemia, and chronic inflammation promote fat accumulation in the liver and the progression of disease in this organ [11]. Overweight and obesity, especially of the abdominal type, are much more common in people with hepatic steatosis [13]. The increase in visceral fat is associated with elevated levels of pro-inflammatory cytokines such as interleukin-6 and tumor necrosis factor α [13]. These cytokines interfere with insulin signaling and inhibit adiponectin secretion, leading to insulin resistance. Such metabolic changes contribute to the development and progression of hepatic steatosis, further accelerating the progression of liver disease in patients with CHC. A study of 324 patients with CHC in the United States found that both obesity and overweight were independently associated with hepatic steatosis [14]. Moreover, multivariate analysis confirmed that hepatic steatosis, especially in advanced stages, is an independent risk factor for fibrosis progression in patients with CHC. Alcohol consumption, even in amounts that do not meet the definition of abuse according to gender, is a known factor that worsens the course of CHC. It increases the risk of serious complications such as cirrhosis, and alcohol-dependent patients, especially women, have a higher risk of death compared to the group without CHC [15]. According to data from a meta-analysis examining the effect of alcohol on liver disease progression in CHC, each drink containing 12 grams of pure alcohol increases the risk of developing cirrhosis by 11% [15]. Alcohol abuse also leads to the development of hepatic steatosis. In a study that analyzed data from the National Health and Nutrition Examination Survey (NHANES), the effect of alcohol consumption was found to be less significant than that of metabolic factors. The increase in the controlled attenuation parameter (CAP) index, used to measure liver steatosis using FibroScan, was statistically significant only in patients who consumed alcohol 5-7 times a week. Frequency of consumption was also more important for the development and progression of steatosis than the amount of alcohol consumed per day [16]. An analysis including a cohort from Spain and the US with liver steatosis associated with metabolic factors showed that alcohol consumption was a factor that independently increased the risk of fibrosis and steatohepatitis. Moreover, already moderate alcohol consumption acted additively with metabolic factors to increase the risk of fibrosis progression. According to the authors, there is no safe daily dose of alcohol [17].
HCV-related liver steatosis
Although metabolic factors play an important role in the development of hepatic steatosis in CHC, viral factors, particularly HCV genotype, are also crucial. Patients infected with GT3 HCV have approximately five times the risk of developing moderate to severe hepatic steatosis compared to those infected with other genotypes [18]. The presence and severity of steatosis in GT3 patients are closely related to viral load [18]. The pathogenesis of HCV-associated hepatic steatosis involves multiple mechanisms affecting lipid metabolism. These include impaired secretion of very low-density lipoprotein (VLDL), increased levels of fatty acid synthase, overregulation of sterol regulatory element-binding protein-1 (SREBP-1), decreased activity of microsomal triglyceride transfer protein (MTTP), and decreased expression of the sterol regulatory element-binding protein-1 (SREBP-1). MTTP reduced expression of peroxisome proliferator-activated receptor α (PPAR-α), and impaired function of phosphatase and tensin homolog (PTEN) [19]. Although GT3 is most commonly associated with these disorders, other HCV genotypes may also contribute to lipid accumulation in the liver. The exact mechanisms underlying HCV-mediated steatosis in infections with other genotypes are not well understood. However, genotype 1 has been shown to be associated with increased expression of suppressor of cytokine signaling-3 (SOCS-3) and tumor necrosis factor alpha, increased levels of reactive oxygen species, decreased adiponectin levels, increased free fatty acids, and impaired fatty acid oxidation due to decreased oxidase activity [10]. Figure 2 presents the main molecular mechanisms by which HCV induces steatosis, depending on the genotype. Liver steatosis is also associated with progression of liver fibrosis. A retrospective study of 603 African-American patients with CHC at Howard University Hospital found that steatosis was an independent risk factor for fibrosis progression (OR: 1.6, p = 0.002), and patients with advanced fibrosis were more likely to have severe hepatic steatosis compared to those with mild fibrosis [20]. Similar observations were reported in a study of 180 patients with CHC [21]. Some studies also suggest that GT3 may be associated with accelerated fibrosis progression [22]. Moreover, a growing amount of evidence points to a link between hepatic steatosis and an increased risk of developing HCC in people with chronic HCV infection [23]. This underscores the importance of monitoring and treating hepatic steatosis as an essential component of strategies to prevent liver disease progression in HCV-infected patients.
Impact of hepatic steatosis on the outcome of CHC treatment
In the era of interferon-based therapy, the presence of hepatic steatosis was considered a significant risk factor for treatment failure in patients with chronic HCV infection [24]. Compared to other well-known predictors of treatment failure, such as viral genotype, the exact impact of steatosis on response to therapy has remained unclear. Studies have shown that insulin resistance may play a role not only in the development of steatosis, but also in treatment failure [12]. An analysis evaluating the effect of insulin resistance and viral factors on achieving a sustained virologic response (SVR) in patients with CHC who were treated with pegylated interferon (pegIFN) and ribavirin showed that the insulin resistance index was an independent predictor of SVR [25]. The percentage of GT1-infected patients with insulin resistance who achieved SVR was lower than in the corresponding group without insulin resistance. In addition, steatosis was associated with a higher rate of relapse in GT3 HCV-infected patients [26]. The analysis, which included more than 400 patients treated with pegIFN and ribavirin for six months, examined the effect of metabolic syndrome on treatment outcomes. It was observed that in the group that met the criteria for a diagnosis of metabolic syndrome, the percentage of patients achieving SVR was lower compared to the group without metabolic syndrome (72.2% vs. 43.7%, p < 0.05) [27]. The introduction of therapies based on direct-acting antivirals (DAAs) has dramatically changed the landscape of HCV treatment [28]. DAAs provide significantly higher SVR rates in a variety of patient populations, rendering many factors that previously influenced treatment failure, including steatosis and insulin resistance, largely irrelevant [28, 29]. Analysis of Polish patients from the EpiTer-2 database showed that although patients with HCV infection and steatosis differed from those without steatosis in terms of body mass index (BMI), genotype distribution, and comorbidities, the effectiveness of DAA therapy was equally high in both groups [30]. In addition, a large cohort study that included more than 8000 German patients did not detect significant differences in treatment efficacy depending on the presence or absence of hepatic steatosis. SVR remained above 96% in both groups, demonstrating that in the era of DAA therapy, steatosis is no longer a significant predictor of treatment failure [31].
Effect of SVR on hepatic steatosis
Direct-acting antiviral therapy and achievement of SVR in patients with CHC is associated with regression of liver fibrosis [32]. However, the effect of HCV elimination on metabolic disorders and hepatic steatosis remains controversial [32]. In a study involving 41 patients following DAA treatment with significant hepatic steatosis, a significant decrease in CAP was observed, accompanied by an increase in low-density lipoprotein (LDL) levels [33]. Increased dyslipidemia may be related to the elimination of HCV particles that bind to lipoproteins during ongoing infection [32]. In contrast, a prospective observational study of Egyptian patients observed increased steatosis one year after DAA treatment, and the presence of steatosis and high BMI were found to be negative predictors of fibrosis regression after therapy [34]. A meta-analysis of 97 original papers that examined changes in metabolic factors, including changes in the severity and presence of hepatic steatosis after DAA treatment, showed a great heterogeneity of results. The inconsistent and relatively short follow-up in the studies that were analyzed also remains a problem [32]. Pre-treatment of existing metabolic disorders, once SVR is achieved, may lead to increased hepatic steatosis and progressive fibrosis, thereby increasing the risk of cirrhosis and HCC despite virus eradication [32]. On the other hand, in patients with genotype 3 infection without metabolic comorbidities, but with hepatic steatosis before therapy, its reduction was observed after achieving SVR [22]. A multidisciplinary approach to patients with metabolic risk factors for hepatic steatosis and an attempt to eliminate them before implementing DAA treatment is needed. The discrepancies in the analyses published to date underscore the need for further research in this area.
Steatotic liver disease and chronic hepatitis B
Epidemiology
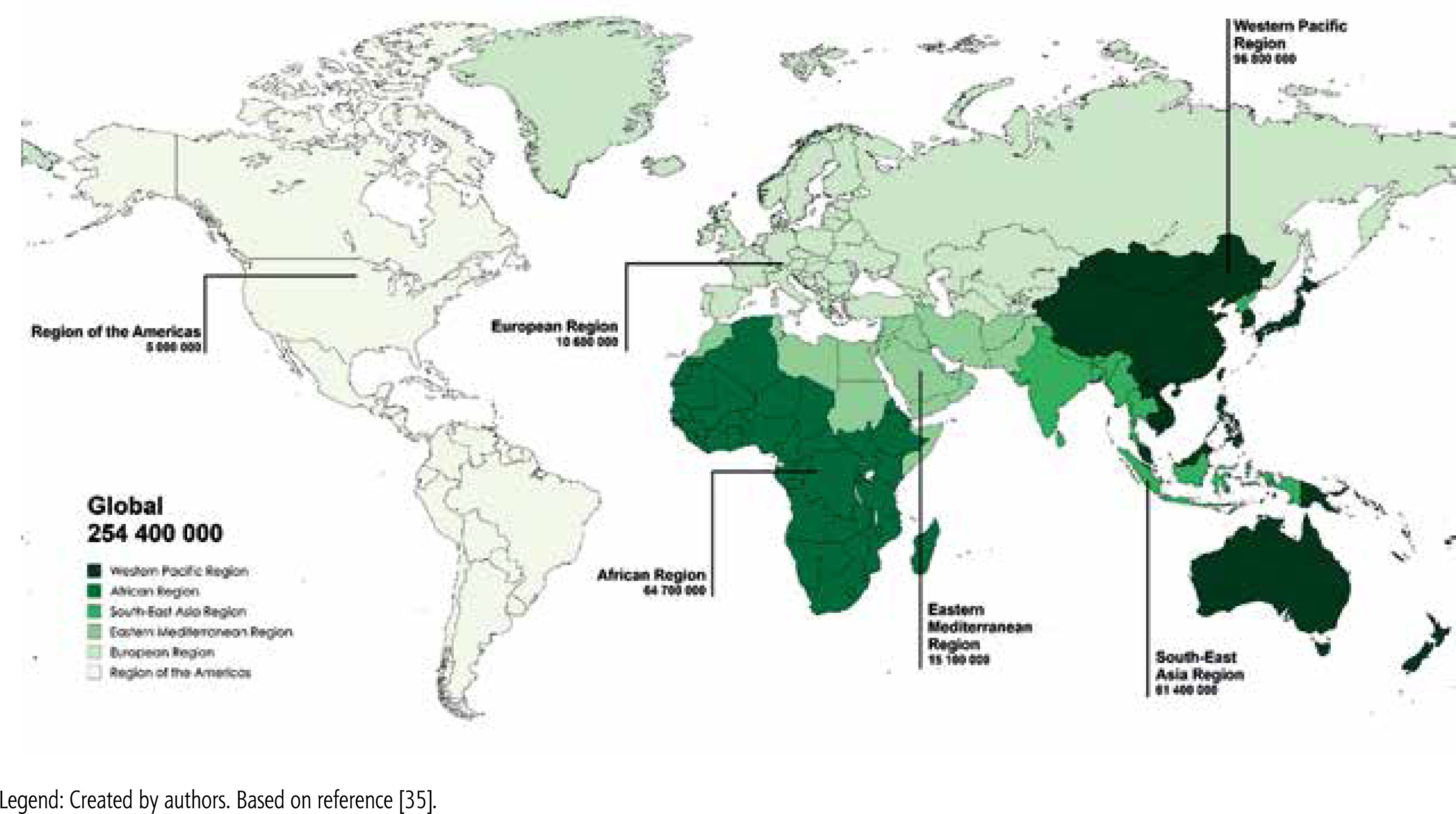
According to a WHO report in 2022, about 254 million people worldwide are chronically infected with HBV, with 1.2 million new infections recorded each year (Fig. 4) [35]. Chronic hepatitis B (CHB) is responsible for about 1.1 million deaths each year, mainly from cirrhosis and HCC [35]. Hepatic steatosis is a common histopathological feature observed in patients with CHB, with a prevalence of 14% to 70%, depending mostly on differences in study populations, diagnostic methods, grading criteria, and genetic or ethnic factors [36]. However, there is no direct evidence to suggest that chronic HBV infection itself increases the risk of hepatic steatosis. The results of available studies indicate that the incidence of hepatic steatosis in patients with CHB is comparable to that in the general population. A recent meta-analysis involving nearly 29 000 patients with CHB found that the incidence of hepatic steatosis was 32.8% (95% CI: 28.9-37.0), which closely mirrors that in the general population [37]. Another meta-analysis, which included 4,100 HBV-infected patients, found an overall SLD prevalence of 29.6% [38], which is consistent with patterns observed in the general population. These findings suggest that hepatic steatosis in patients with CHB is mainly due to metabolic factors rather than HBV infection itself. Notably, several studies have reported an inverse relationship between HBV DNA levels and hepatic steatosis, indicating a protective effect of HBV infection on hepatic steatosis [39]. In patients with CHB and concomitant SLD, a progressive decrease in median HBV DNA levels was observed as the severity of steatosis increased. Interestingly, higher degrees of steatosis were independently associated with lower serum HBV DNA levels, suggesting that steatosis may have a suppressive effect on HBV replication [40].
Risk factors associated with the development of hepatic steatosis and progression of liver disease in patients with CHB
The development of hepatic steatosis in patients with CHB, as in the general population, appears to be directly related to the presence of metabolic risk factors, such as central obesity, unfavorable lipid profile and diabetes. Steatosis can develop in any age group, but because the prevalence of metabolic risk factors increases with age, patients with CHB and SLD tend to be older than patients in control groups [41, 42]. Due to the protective role of estrogen in women, which delays the development of SLD, gender also plays an important role [41, 42]. In a meta-analysis aimed at analyzing factors affecting the development of steatosis in patients with SLD, both male sex and older age were found to be positive predictors of the presence of SLD. Similar results were found for metabolic risk factors [37]. In a case-control study in which groups were matched for age, gender, treatment status, and duration, among patients with CHB and SLD who had not received prior treatment, 80% had at least one metabolic risk factor for developing steatosis. Moreover, regardless of treatment status, both BMI and obesity independently increased the risk of developing SLD. In the untreated group, the presence of steatosis was also considered a predictor of a metabolic syndrome [40]. The influence of metabolic risk factors on the development of SLD in patients with CHB, independent of the virological component, was confirmed in a study in which more than 30 000 patients were included, and more than 9% were infected with HBV. Regardless of virological status, patients with SLD were predominantly male, older, and characterized by central obesity, higher blood pressure, and impaired lipid metabolism. In multivariate analysis, metabolic risk factors independently increasing the risk of developing SLD were similar in both patients with HBV and in the uninfected group [42].
Interaction between hepatic steatosis and CHB
The interaction between chronic HBV infection and SLD is complex and is the subject of ongoing research, with many reporting that hepatic steatosis can inhibit HBV replication. SLD is associated with increased immune activity, including higher expression of Th17 cell-related genes and increased levels of interleukin 21, leading to production of inflammatory cytokines, decreased HBV replication, and clearance of HBV DNA and HBV viral e antigen (HBeAg) [43]. In addition, SLD-induced metabolic stress can lead to inhibition of HBV replication and apoptosis of HBV-infected cells, helping to clear the virus and limit the progression of HBV-related disease [43]. However, despite lower HBV replication, patients with CHB and SLD often experience faster progression of liver disease compared to patients with only CHB or SLD [44]. The synergistic effect of chronic HBV infection and SLD in driving liver disease progression is often explained by the “two-strike theory”. In this model, viral infection serves as the first hit, causing initial liver damage, while steatohepatitis serves as the second hit, exacerbating inflammation and fibrosis and accelerating disease progression. HBV HBx protein stimulates genes associated with lipid accumulation, such as PPAR, SREBP and FABP1, promoting steatosis and hepatocyte proliferation while suppressing apoptosis, contributing to the development of HCC [45]. In addition, oxidative stress associated with SLD can create a pro-fibrotic and pro-carcinogenic environment, further promoting carcinogenesis [45]. Studies from daily clinical practice involving large patient populations have confirmed higher rates of liver cancer and mortality in patients with CHB coexisting with SLD. A study in Hong Kong found a 7.3-fold increase in liver cancer risk in patients with CHB and SLD [46]. Similarly, Lee et al. found that concurrent hepatic steatosis in patients with CHB was associated with a higher risk of HCC [47].
Management of CHB-SLD patients
The treatment of SLD in patients with CHB requires a comprehensive and multifaceted approach. Non-invasive measurements, such as elastography with CAP assessment using FibroScan, can effectively assess both liver fibrosis and steatosis and nowadays represents a valuable alternative to histopathological examination, which in the past was the gold standard for determining the etiology and severity of liver disease [48]. Antiviral treatment remains key in the treatment of CHB, and nucleos(t)ide analogues (NAs) such as tenofovir and entecavir are effective in inhibiting HBV replication [49]. The decision to implement antiviral therapy in patients with chronic HBV infection is based on the current criteria contained in the recommendations of international and national scientific societies taking into account three parameters: alanine aminotransferase activity, HBV viral load, and the stage of fibrosis in the liver [49–51]. To date, no drugs have been registered to treat MASH with fibrosis in Europe, but ongoing clinical trials, many of which are already in Phase 3, offer hope that such forms of treatment will be available in the near future [52]. The first candidate appears to be resmetirom, an oral thyromimetic drug approved in March 2024 by the US Food and Drug Administration for the treatment of MASH in patients without cirrhosis [53]. Thus, for the time being, lifestyle modification remains the standard of care for a patient with SLD, including a patient with concomitant CHB. Patients who are overweight or obese are advised to lose weight, in particular to reduce 3-5% of total body weight to improve steatosis and 7-10% to obtain MASH resolution [54]. Patients with diabetes and arterial hypertension are recommended to effectively treat these conditions under the supervision of medical specialists. The results of analyses on the effects of antiviral treatment for patients with CHB and SLD are inconsistent; some studies show lower virological response rates, while others show no difference compared to patients without SLD. A recent meta-analysis involving 48,472 patients found that coexisting hepatic steatosis was negatively associated with response to antiviral treatment in patients with SLD [55]. Similarly, Jin et al. identified hepatic steatosis as an independent factor contributing to treatment failure with entecavir [56]. However, other studies have reported no significant differences in outcomes between CHB patients with and without SLD treated with NAs [57]. Taken together, these results underscore the importance of regular monitoring of disease progression in patients with CHB and SLD.
Summary
The co-occurrence of SLD with chronic hepatotropic virus infection is comparable to the prevalence of SLD in the general population for HBV and higher for HCV. In addition to the contribution of classic patient-related metabolic risk factors such as obesity, diabetes and hypertension, virus-related factors such as HCV genotype influence disease progression. The coexistence of SLD with chronic viral hepatitis can exacerbate liver damage, accelerate fibrosis progression, and increase the risk of developing cirrhosis, while data on the impact on the effects of antiviral therapy are inconclusive.





