

Introduction
Activated phosphoinositide 3-kinase δ syndrome (APDS) is a primary immunodeficiency disorder (PID) that was initially described in two independent studies in 2013 and 2014 [1, 2]. It can manifest in three forms – APDS type 1, APDS type 2, and APDS-like (APDS-L) – each caused by different monoallelic germline variants in three genes related to the phosphatidylinositol 3-kinase (PI3K) signaling pathway. This pathway plays a crucial role in cell proliferation and maturation. Phosphoinositide kinase class δ (PI3Kδ) is predominantly expressed in immune cells. APDS 1 is caused by various gain-of-function (GOF) mutations in the PIK3CD gene, which encodes the catalytic subunit p110δ of the PI3Kδ enzyme complex. APDS 2 results from loss-of-function (LOF) mutations in the PIK3R1 gene, encoding the regulatory subunit p85α of PI3K enzyme complexes, including PI3Kδ. APDS-L arises from a LOF variant in the PTEN gene, which encodes PTEN phosphatase – a molecule with an activity opposite to PI3K [3]. In all cases, these variants lead to excessive activation of the PI3K signaling pathway, resulting in immunological system disorders.
Among the predominant clinical presentations of APDS are recurrent respiratory tract infections including pneumonia, bronchiectases, lymphadenopathy, hepatosplenomegaly, autoimmune diseases, neurodevelopmental delay and an increased risk of lymphoma [4].
Patients with APDS typically exhibit hypogammaglobulinemia alongside features of hyper-IgM syndrome (HIGM), lymphopenia in B- and T-cell lines as well as elevated levels of T CD8+ effector memory cells, leading to low CD4+/CD8+ index values [4].
Approximately 28% of APDS patients suffer from autoimmune diseases with hematologic manifestations such as autoimmune hemolytic anemia (AIHA) and immune thrombocytopenic purpura (ITP), which together make up 76% of autoimmune disorders observed in APDS [5].
The diagnostic process of APDS is challenging, with a median delay of 7 years before a proper diagnosis is established. Common primary clinical misdiagnoses include HIGM, lymphoma, common variable immunodeficiency (CVID) and combined immunodeficiency (CID). Around 2.6% of patients are initially misdiagnosed with autoimmune lymphoproliferative syndrome (ALPS). The prevalent initial clinical manifestation is usually respiratory tract infection, while autoimmunity presents as the first clinical symptom in only 1.8% of patients [5].
The treatment options available for APDS include antibiotic prophylaxis, immunoglobulin infusions, immunosuppressive or selective PI3Kδ inhibitors and hematopoietic stem cell transplantation (HSCT) [6]. HSCT represents an effective therapy associated with a 2-year overall survival (OS) rate of 86%. However, it is also associated with a high incidence of infectious, immunological and toxic complications [7].
Case description
A 6-year-old boy with a history of recurrent infections, multiple episodes of lymphadenopathy and hepatosplenomegaly, diagnosed with APDS 1, was admitted to our clinic for HSCT.
In the first weeks of the patient’s life, frequent and intense vomiting after feeding began. The diagnostic process at that point revealed normocytic anemia with reticulocytosis, leukopenia and thrombocytopenia, along with hepatomegaly. At the age of four months, he suffered from his first respiratory-syncytial virus (RSV) pneumonia, leading to anemia requiring a blood transfusion. Over the next six years, the patient continuously suffered from recurrent viral and bacterial respiratory tract infections including pneumonia as well as gastrointestinal and soft tissue infections often accompanied by anemia. Key clinical findings were repetitive hepatosplenomegaly and lymphadenopathy of mesenteric, periaortic, mediastinal, cervical, and axillary nodes. Ultrasound examination also revealed hypoechogenic multifocal lesions in the thyroid, submandibular and sublingual glands. No neurodevelopmental delay was observed.
In the meantime, the diagnostic investigation continued. Major events are chronologically presented in Figure 1. Negative Coombs test ruled out an autoimmune etiology of anemia. Even though hematological anomalies and simultaneous lymphadenopathy raised the suspicion of juvenile myelomonocytic leukemia (JMML), genetic testing did not reveal the presence of JMML-associated mutations (PTPN11, KRAS, NRAS, c-CBL). Bone marrow biopsies showed no evidence of malignancy. Furthermore, tests for alpha- and beta-thalassemia were negative. Due to the suspicion of Langerhans cell histiocytosis (LCH) (seborrheic lesions on the head) a skin biopsy was taken yet histopathological examination did not confirm LCH. Enzymatic tests excluded Gaucher and Niemann-Pick disease. Additionally, abdominal biopsies of lymph nodes and the large intestine did not indicate malignant proliferation. Periodic observation of scant viremia was seen in CMV-PCR and EBV-PCR. The presence of antiplatelet antibodies was confirmed. The cross-sectional laboratory results along with lymphocyte subpopulations are presented in Table 1.
Fig. 1
Timeline illustrating major clinical events or findings (above the arrow) and some relevant steps in the diagnostic process (under the arrow). GI – gastrointestinal, JMML – juvenile myelomonocytic leukemia, LCH – Langerhans cell histiocytosis, ALPS – autoimmune lymphoproliferative syndrome, APDS – activated phosphoinositide 3-kinase δ syndrome
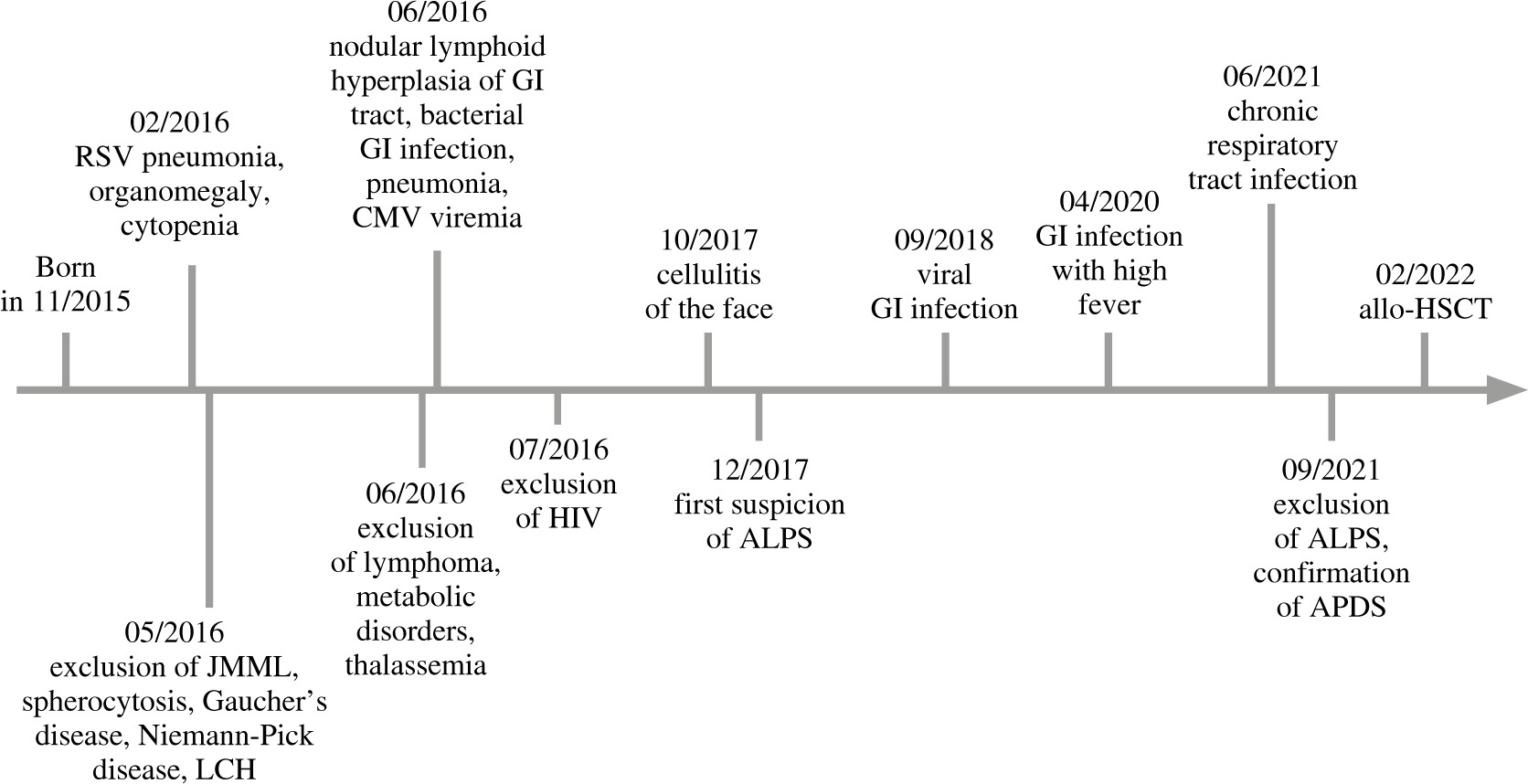
Table 1
The cross-sectional laboratory outcomes (obtained from our patient at the age of five) and lymphocyte subpopulations (obtained from our patient at the age of three). Given B cell count comes from laboratory testing conducted during the 5th year of life because its examination at the time of obtaining a detailed lymphocyte profile did not reveal any abnormality in B cell quantity. The presented data illustrate some typical changes seen in APDS including thrombocytopenia, a marked increase in IgM concentration, a low CD4/CD8 index, skew towards effector memory cells and a decrease in B lymphocyte count. These anomalies were periodically observed throughout the patient’s life. The reference values for the absolute size of T-helper and T-cytotoxic cell subpopulations are calculated based on the relative subpopulation size reference values provided by the laboratory. SSC – side scatter
The clinical presentation led to further diagnostics for an immunodeficiency disorder, revealing decreased numbers of both CD4+ and CD8+ lymphocytes, along with a low CD4+/CD8+ index and elevated IgM level. Screening tests for HIV infection were negative. In the initial testing for suspected ALPS a heterozygotic variant of caspase 10 was found. However, subsequent diagnostic panels based on next-generation sequencing (NGS) excluded ALPS-causing variants and instead revealed two missense mutations (V888M and E1021K) in the PIK3CD gene. Further genetic testing of the patient’s parents using the Sanger method demonstrated that his mother carried the V888M mutation but remained asymptomatic. No specific mutation was found in the patient’s father.
Nearly 6 years after the onset of the first symptoms, the patient was diagnosed with APDS 1. Due to the recurrent infections, lymphadenopathy, organomegaly and the unavailability of selective PI3Kδ inhibitors, as well as the high risk of lymphoma, he was eligible for allogeneic matched unrelated donor HSCT and was referred to our clinic.
Upon admission, the patient was in a good general condition, showing no signs of infection. Routine clinical examination did not reveal any abnormalities, and his psychomotor development was appropriate for his age.
Peripheral blood stem cell transplantation (PBSCT) from an unrelated, HLA high-resolution 10/10 allele-matched, AB0-identical donor was performed. Conditioning included treosulfan 36 g/m2, fludarabine 5 × 30 mg/m2 and thiotepa 2 × 5 mg/kg. As a serotherapy anti-thymocyte globulin (Grafalon) 3 × 15 mg/kg was administered on days –3 to –1. Cyclosporine A from day –1 and methotrexate 10 mg/m2 on days +1, +3, +6 were used as graft-versus-host disease prophylaxis. The PBSC graft contained 5.02 × 106 CD34+ cells/kg. From day +7 G-CSF was administered.
During the peritransplant period, the patient suffered from several complications, including oral mucositis, CMV reactivation, ESBL+ Escherichia coli gastrointestinal infection, arterial hypertension and acute graft-versus-host disease (aGvHD) classified as grade II. However, all these complications were successfully managed using conventional measures.
Neutrophil engraftment (defined as the first of three consecutive days after achieving a sustained peripheral blood neutrophil count of > 0.5 × 109/l) occurred at day +13. Platelet engraftment (defined as the first of seven consecutive days after achieving a sustained peripheral blood platelet count of > 20 × 109/l) occurred at day +27.
Post-transplantation blood testing during the first months after transplantation showed stable, full donor chimerism. Subsequent donor cell infusions were not administered. However, one year after HSCT, the emergence of a 12% autologous signal in peripheral blood mononuclear cells (MNC) chimerism was observed. Due to a persistently low B-lymphocyte count and decreased levels of immunoglobulins, the patient has been receiving immunoglobulin substitution. Blood count, lymphocyte subpopulations, immunoglobulin concentrations and chimerism in the peri- and post-transplant period are presented in Table 2 (reference values for lymphocyte subpopulations come from the study by Comans-Bitter et al. [8]).
Table 2
Laboratory outcomes of the patient before and after HSCT. Reference values for lymphocyte subpopulations come from the study by Comans-Bitter et al. [8]. WBC – white blood count, HGB – hemoglobin concentration, PLT – platelet count, NEU – neutrophil count, LYMPH – lymphocyte count, NA – not applicable, ALLO – fully allogeneic chimerism – autologous signal not detected, AUTO – percentage of autologous signal
After a 17-month-long post-HSCT follow-up, the patient continues to maintain good general health despite occasional and mild episodes of infections. However, due to mixed chimerism and dependence on immunoglobulin substitution, he remains under surveillance.
Discussion
We have presented a case of successful treatment of an APDS 1 patient with matched unrelated donor HSCT.
According to data collected by Jamee et al. [5], the median delay in the diagnosis of APDS patients is approximately 7 years. In the case of our patient, symptoms started in the first weeks of life, and he was diagnosed with APDS when he was nearly 6 years old. Throughout the diagnostic process, several potential diagnoses were considered but later excluded, as listed in Table 3. Therefore, APDS should be considered in the differential diagnosis of a wide range of diseases, including malignancies, metabolic disorders, autoimmune conditions and other immunodeficiencies.
Table 3
Chronologically listed diseases suspected and excluded during the diagnostic process of the patient. APDS should be considered during differential diagnosis of these entities
Although APDS is primarily described as a PID, it can have other manifestations which precede the onset of infective complications in approximately half of the patients [5]. One of the first signs of the disease observed in our patient was tree-lineage cytopenia. The autoimmune nature of thrombocytopenia was confirmed by detecting antiplatelet antibodies. Autoimmunity as the first sign of APDS is rare. Moreover, in the group described by Jamee, the two patients with autoimmunity as the first sign were much older than our patient [5]. The atypical chronology of symptoms and uncharacteristic primary symptoms led to a 6-year delay in diagnosis. This case highlights the significance of increasing awareness about APDS.
Autoimmune etiology of our patient’s anemia was excluded. While some reviews by Redenbaugh and Coulter [3] and Thouenon et al. [9] discuss anemia in APDS as a result of autoimmunity, it could result from other mechanisms. Seidel [10] categorized causes of cytopenias in PIDs. In the case of our patient, possible causes of anemia were lymphoproliferation, splenomegaly or chronic viral infection. A similar etiology could be assigned to recurrent leukopenia. Regardless of whether the cause of cytopenia is autoimmune or not, it is considered a major and early feature of APDS. Low hemoglobin levels were observed in the majority of APDS patient cohorts described by Qiu et al. [11]. Miano et al. [12] described a case of a 1-year-old patient with a novel variant of the PI3KCD gene, and pure red cell aplasia associated with AIHA. Rivalta et al. [13] reported on APDS patients with chronic EBV infection who suffered from episodes of hemolysis in early childhood. Thus, cytopenia even without an apparent infection can be an indicative sign of APDS.
Abnormalities in lymphocyte subsets are common features of APDS. The most frequent anomalies include increased transitional B-cell counts, reduced class-switched memory B-cell counts, reduced naive T-cell counts and increased effector CD8+ T cells [14]. Although we did not obtain the patient’s B-cell subpopulation counts, his T-cells presented the typical skew towards effector memory cells [15], as shown in Table 1. Other common features of APDS are hypogammaglobulinemia and elevated IgM. Despite normal values of IgG immunoglobulin concentration, our patient’s IgM levels were markedly elevated, as depicted in Table 1.
The patient presented two variants in the PIK3CD gene. E1021K is the most frequent variant in APDS 1 [5], but V888M, which was also detected in the patient’s asymptomatic mother’s genome, should not be interpreted as a contributing factor.
Recurrent EBV and CMV viremia are widely described features of APDS, occurring in approximately 26% and 15% of patients, respectively [16]. Rivalta et al. [13] reported on 4 patients with chronic EBV replication, two of whom suffered from diffuse large B-cell lymphoma. Yin et al. [17] described the first occurrence of plasmablastic lymphoma in a 6-year-old APDS patient who previously presented with recurring EBV infections. These findings, along with the review conducted by Riaz et al. [18], indicate that both chronic EBV viremia and presence of the APDS variant are risk factors of malignancy. Taking these data into consideration, HSCT is a well-justified treatment option for APDS patients.
Continuously observed lymphadenopathy or lympho- proliferation with various localizations (abdominal lymph nodes, intestine wall, thyroid, sublingual, and submandibular glands), accompanied by splenomegaly, without evidence of malignancy, raised suspicion of ALPS. The main symptoms of ALPS include autoimmune cytopenia, lymphadenopathy, hepatosplenomegaly, and various skin lesions [19]. In particular, the early symptoms of our patient, with cytopenia, splenomegaly, and lymphadenopathy, were suggestive of ALPS. However, in a long-term study presented by Tessarin et al. [15], seven out of eight APDS 1 patients had benign multifocal lymphoproliferation and four of them started diagnostics due to this primary diagnosis. Such observations suggest that lymphoproliferation in APDS should be viewed as a hallmark, as it is in ALPS.
Several descriptive studies refer to outcomes of HSCT in APDS. One of the first reports, presented by Imai et al. [20] described six transplanted patients, four of whom were successfully treated by HSCT. Okano et al. [21] in a retrospective study with a remarkable follow-up reported nine transplanted APDS patients with 30-year OS of 86%. This result seems similar to the outcomes of HSCT in other PIDs [22]. Coulter [16] reported five cases of HSCT in APDS. Three patients were successfully transplanted, one suffered poor engraftment, resulting in the need for long-term immunoglobulin therapy, and one patient died two years after the procedure (sepsis). Nademi et al. [23] presented eleven patients undergoing HSCT, nine of whom stayed alive with follow-up between 8 months and 16 years.
Dimitrova et al. [7] described the largest (57 patients) cohort of APDS patients treated with HSCT. None of these patients were transplanted preemptively. Indications for HSCT included hematologic malignancy (26%), nonmalignant lymphoproliferation (49%), autoimmune cytopenias (14%), chronic or recurrent infections (46%), and end organ damage (42%). The 2-year OS was 86%. Transplant-related mortality occurred due to infections (11% of patients) or organ-related regimen toxicity (4%).
The above-mentioned reports present similar outcomes, suggesting that HSCT should be considered for patients with APDS who are unresponsive to other treatment therapies or do not have access to novel treatments such as selective PI3Kδ inhibitors, as it was in the case of our patient. Tessarin’s study [15] also came to similar conclusions regarding HSCT as a treatment option for APDS. The decision for HSCT was prompted by the high risk of lymphoma and poor symptom control.
The importance of allo-HSCT in the treatment of APDS was discussed in comparative description of two Polish patients, one with APDS 1 and the other with APDS 2 [24]. Both patients suffered from multiple infections and lymphadenopathy. Both of them were initially treated with immunoglobulin replacement therapy. Only the patient with APDS 2, who initially had less widespread lymphoproliferation and milder lymphocyte profile deviations, showed clinical improvement after immunoglobulin therapy. The APDS 1 patient, resistant to the treatment, underwent successful allo-HSCT from his healthy, histocompatible brother, with a good clinical effect. The severity of his symptoms, along with presence of CMV viremia, which increases the risk of malignancy, and the final treatment appear to be similar to our patient’s case.
In Dimitrova’s study, 75% of patients had APDS 1 [7]. Similarly, in the study reported by Nademi [23], APDS 1 patients constituted the majority of cases. However, there were no significant differences in OS and graft failure-free survival (GFFS) between APDS 1 and APDS 2 patients. The difference in the number of patients undergoing HSCT in the APDS 1 and APDS 2 groups may result from the differences in the overall occurrence of the two APDS types or from the fact that APDS 2 patients might respond better to alternative therapies. We observed a positive response to the conservative treatment regimen in a patient with APDS 2 who received immunoglobulins and remained in a good general condition. This patient has been described by our colleagues from Poznan [25].
The conditioning regimen preferred in PID patients is reduced toxicity conditioning (RTC) [26]. In Dimitrova’s cohort, RTC was the most frequent approach and it showed higher OS and GFFS compared to other strategies, although the difference was not statistically significant. The most frequently examined approach among RTC regimens in PID patients was the combination of treosulfan and fludarabine (TREO/FLU), which could be extended by addition of thiotepa. However, addition of thiotepa to the TREO/FLU regimen did not result in any relevant difference, as reported by Dinur-Schejter [27]. Treosulfan was mainly administered at a dose of 42 g/m2; however, the doses should be reduced to a total of 36 g/m2 in patients weighing less than 12 kg [28]. Our patient weighed 12 kg, and thus he received RTC with a reduced dose of treosulfan, a standard dose of fludarabine and addition of thiotepa.
The incidence of acute GvHD after HSCT in APDS or PID patients varies among studies. Vora et al. [29] reported grade > II acute GvHD in 67% of 69 PID patients treated with HSCT, but among a cohort of 39 transplanted PID patients described by Ballantyne [30] only 5% developed grade > II acute GvHD. In Nademi’s cohort [23], GvHD occurred in 81%, whereas Dimitrova [7] reported a 39% incidence of GvHD with a maximum grade of III. These findings suggest that GvHD is a common complication after HSCT in APDS, although its precise incidence is difficult to evaluate.
Considering the median times in the cohort described by Dimitrova and Nademi, our patient achieved neutrophil and platelet engraftment in a predictable period. Also the dynamics of our patient’s CD3+CD8+ and CD3+CD4+ cell concentrations were comparable with the outcomes of Dimitrova’s cohort [7, 23]. Surprisingly, we observed rapid reconstitution in the NK cell population, which was seen only in a few cases of Dimitrova’s study. Recovery in the B-cell lineage took longer (requiring immunoglobulin substitution) and together with still increasing mixed chimerism (MNC), there is a slight risk of secondary graft failure. Dimitrova et al. reported mixed chimerism in 17% of phenotype-reversed patients, with the highest incidence occurring between the twelfth and twenty-third month after HSCT. However, the percentage of donor chimerism for all patients increased after this period [7]. Our patient’s course was complicated by mixed chimerism in predictable periods, and he still has a chance of complete recovery.
This case report contributes to the process of expanding the use of HSCT as a therapeutic approach in APDS. Such reports are meant to increase the knowledge about the efficacy and safety of transplantation in this PID. The recently reported successful haploidentical HSCT in an APDS 1 patient [31] further extends therapeutic possibilities for patients suffering from this condition.
Conclusions
The presented literature, alongside our case, demonstrates the difficulty in the diagnosis of APDS and confirms that HSCT is a challenging yet effective treatment method for this condition. APDS should be considered in the differential diagnosis of various disorders including malignancies, metabolic and autoimmune diseases as well as other immunodeficiencies. Raising awareness of APDS is crucial to reduce the diagnostic delay. HSCT with RTC and serotherapy might be an optimal approach for pediatric patients with APDS when conservative treatment proves ineffective. Moreover, it may reduce the high risk of malignancy. However, post-HSCT patients still require close monitoring.