

Introduction
Chimeric antigen receptors (CARs) have been used to redirect the specificity of autologous T-cells against leukemia and lymphoma with promising clinical results [1-3]. Thus, CAR-T has been become a major breakthrough in cancer therapy, especially in treatment of hematological malignancies. However, therapy based on CAR-T has drawbacks including high manufacturing costs, a significant risk of graft-versus-host disease (GVHD) and potentially fatal toxicities such as cytokine release syndrome (CRS) [4]. Theoretically, NK cells, which represent another important cell type for adoptive immunotherapy, may be an alternative driver for CARs. CAR-NK cells are considered safer than CAR-T cells due to their short lifetime and production of lower levels of toxic cytokines, including interferon γ (IFN-γ) and granulocyte-macrophage colony-stimulating factor (GM-CSF), than those produced by T cells [5]. Additionally, as activating NK cells do not require strict HLA matching, the CAR-NK therapy does not carry a risk of GVHD.
Unlike primary NK cells, which are difficult to isolate, purify and transduce, the human NK cell line NK-92 can be continuously expanded in the presence of interleukin (IL)-2 in a Good Manufacturing Practice-compliant process [6]. The cytolytic effects and safety of NK-92 have been confirmed in preclinical studies and clinical studies [6, 7]. Presently both primary NK cells and the NK-92 cell line are used to generate CAR-NK cells targeting several different antigens, including CD19, CD20, CD33, CD138, CD3, CD5, CD123 and SLAMF7 [8]. CD19-directed CAR-NK-92 showed increased cytotoxic activity against leukemia cells expressing CD19 [9]. Apart from NK-92, there are six other known NK cell lines, while none of these showed consistently and reproducibly high antitumor cytotoxicity. Additionally, only NK-92 cells can easily be genetically manipulated to recognize specific tumor antigens and be infused to cancer patients with clinical benefit and minimal side effects [10].
B cell lymphoma is a heterogeneous group of diseases that includes non-Hodgkin (90%) lymphoma and Hodgkin lymphoma (10%) [11]. Though advances in therapy have substantially increased the likelihood of cure for patients with B cell lymphoma, outcomes have been poor for patients with relapsed/refractory disease. After 2 or more lines of therapy, CAR-T cell therapy offers a potentially curative therapy to previously incurable patients. At present, the most popular target of CAR-T therapy for B cell lymphoma is CD19. CD19 CAR-T is widely used to treat B-cell-derived malignancies, including B-acute lymphatic leukemia (B-ALL), B-chronic lymphatic leukemia (CLL) and B-non-Hodgkin’s lymphoma (B-NHL). Though an encouraging outcome emerged when anti-CD19 CAR-T products were used in several subtypes of B cell lymphoma, progression or relapse appeared eventually in the majority of patients after anti-CD19 CAR T-cell therapy [12, 13]. One main factor leading to anti-CD19 CAR-T failure is that tumor cells can evade the recognition and attack by CD19 CAR-T through downregulating or even losing surface CD19 expression. A study showed that about 10% of B-ALL patients relapsed after CD19-targeted CAR-T therapy due to antigen loss [14]. Thus, there is a great need to seek alternative targets for immunotherapy of B cell lymphoma in case antigen loss occurs.
CD22 is a sialic acid-binding adhesion molecule expressed on most B cell lymphoma patients [15, 16], while its expression on normal tissue is restricted to the B cell lineage. It is reported that monoclonal antibody (mAb)-based therapeutics targeting CD22 have attained promising results in clinical trials [17-19]. There was a study showing that CD22-CAR expressing T cells can mediate similarly potent antineoplastic effects as CD19 CAR-T [20]. In the present study, we accomplished generation as well as molecular and functional characterization of a clonal CD22-redirected CAR-NK-92 cell line. We also detected whether the cytolytic potency could be influenced after irradiation and the expression level of inhibitory receptors after repeated antigen stimulation. In summary, our results suggested that adoptive transfer of CD22-redirected CAR-NK-92 cells may serve as a promising strategy for treatment of B cell lymphoma.
Material and methods
Cells and culture conditions
The three lymphoma cell lines Raji, TMD8 and Daudi, which were all CD22 and CD19 positive, were cultured in RPMI 1640 medium (Gibco). 293T cells were used for the lentivirus and retrovirus vector packaging cell line (ATCC) and cultured in DMEM medium (Gibco). All the above cell culture medium was supplied with 10% heat-inactivated fetal bovine serum (FBS; Biological Industries), 100 U/ml penicillin, 100 µg/ml streptomycin, and 2 mM L-glutamine (Invitrogen). The NK-92 cell line was cultured in α-MEM medium (Gibco) supplemented with 12.5% heat-inactivated FBS (Biological Industries) and 12.5% heat-inactivated Horse Serum (Gibco) in the presence of recombinant human IL-2 (teceleukin, rhIL-2; Roche) at the final concentration of 500 IU/ml.
Construction of CAR NK-92 cells
The single chain variable fragment (scFv) m971 to target CD22 originated from a fully humanized anti-CD22 antibody [21]. To generate anti-CD22 CAR expression constructs, the fragments encoding the CD8 leading peptide, anti-CD22 scFv (m971), CD8 hinge and 41BB-CD3ζ were synthesized by Genewiz (Suzhou) and were amplified by PCR. Then these fragments were cloned into the pCDH-SFFV-IRES.EGFP lentiviral plasmid using EcoR I and Not I sites. The resultant plasmid was designated pCDH-SFFV-m971-41BB-CD3ζ-IRES.EGFP. The methods for production of lentivirus particles encoding CAR and transduction of cells have been described in our previous study [22]. The expression plasmids together with the packaging plasmids including psPax2 and PMD2.G were cotransfected into 293T cells at a ratio of 20 µg : 15 µg : 6 µg, and 48 hours after transfection, the collected supernatants were filtered. NK-92 cells were resuspended in the virus solution in the presence of 500 IU/ml IL-2 and 4 µg/ml polybrene (Sigma) and centrifuged at 600 g for 45 minutes, and then transferred into a CO2 incubator. 8 hours later, the virus solution was replaced with fresh NK-92 medium (mentioned above). After culture for 72 hours, EGFP-positive cells were sorted as CAR-expressing cells by flow cytometric cell sorting with a FACS Melody cell sorter (BD Biosciences).
Flow cytometry analysis of CAR expression
The surface expression of CAR on NK-92 cells was measured by flow cytometry. CAR-transduced NK-92 cells were resuspended with wash buffer (1 × PBS + 1% BSA), and incubated with 1 µg/ml biotinylated protein L (Thermo Scientific) or normal polyclonal goat immunoglobulin G (IgG) as an isotype control. The cells were then incubated with biotin-streptomycin-APC (BD Bioscience) and analyzed by flow cytometry. Data analysis was carried out using FlowJo software (Tree Star Inc.).
Immunoblotting
The sorted NK-92 cells after lysis were separated by SDS-PAGE gel and then transferred to the PVDF membrane (Millipore). The membranes were incubated with anti- human CD247 (CD3ζ) primary antibody (BD Pharmingen). Then membranes were washed and developed using a secondary anti-mouse IgG-HRP antibody (Biworld). Finally, the expression of total CAR protein in NK-92 cells was detected by an enhanced chemiluminescence reagent (GE Healthcare Biosciences).
Generation of lymphoma cell lines stably expressing firefly luciferase
Lymphoma cell lines stably expressing firefly luciferase were generated through transduction with lentiviruses encoding firefly luciferase. EGFP positive cells were sorted as the firefly luciferase expressing cells.
Cytotoxicity assay
Mock NK-92 cells or CAR NK-92 cells were cocultured with three lymphoma cell lines (2 × 104) expressing firefly luciferase at the indicated effector/target ratios (E/T) in the 96-well U-bottom plate at 37oC for 6 hours. Target cells alone were also cultured as the control. At the end of the co-culture, the Bright-Glo luciferase reagent (Promega) was added into the wells directly, and the luminescence value was recorded using a luminescence microplate reader (BioTek). Then the bioluminescence value was detected with the substrate of firefly luciferase. The percentage of specific lysis was calculated as: (the luminescence values of target cells alone – the luminescence values of target cell cocultured with T cells)/the luminescence values of target cells alone × 100%.
For the detection of NK cell-mediated lysis of patient lymphoma cells, primary lymphoma cells prelabeled with eFluor 670 (eBioscience) were cultured alone or cocultured with NK cells for the indicated time period. 123 count beads (eBioscience) were added and the absolute number of eFluor 670-labeled target cells was determined by flow cytometric analysis according to the manufacturer’s protocol. The percentage of specific lysis was calculated as: (the absolute number of target cells when cultured alone – the absolute number of target cells when cocultured with T cells)/the absolute number of target cells when cultured alone × 100%.
Cytokine release assays
Effector cells (2.5 × 105) were cocultured with target cells at an E/T ratio of 1 : 1 in 96-well U-bottom plates at 37oC for 24 hours, and then the cell-free supernatants were collected for the IFN-γ and tumor necrosis factor α (TNF-α) secretion detected by the method of ELISA kits (R&D system) according to the manufacturer’s protocol.
CD107a degranulation assay in the NK-92 cells
Effector cells (3 × 105) were cocultured with the same number of target cells in 96-well U-bottom plates for 4 hours at 37oC. In addition, mock NK-92 cells or CAR NK-92 cells were incubated without target cells for the negative control. After 2 hours, monensin and APC conjugated anti-CD107a antibody (BD Biosciences) or IgG1 isotype antibody (BD Biosciences) were added. The cells were further stained with PE-Cy7-conjugated anti-CD56 antibody (BD Biosciences) and analyzed with flow cytometry.
Irradiation of NK-92 cells
m971 BBZ and mock NK-92 cells were collected by centrifugation, counted, washed, resuspended in fresh medium and irradiated with 5 or 10 Gy using a X-RAD320ix device (PXI Systems, USA). For in vitro cytotoxicity and cytokine release assay, irradiated cells were washed, resuspended in fresh growth medium and cultured for up to 8 days, then used for the subsequent assays.
Repeated stimulation of effector cells
Effector cells including mock, m971-BBZ NK-92 and FMC63 BBZ T cells were co-incubated with target cells (TMD8) for 3 days at an initial E/T ratio of 1 : 2 in 24-well plates. Fresh target cells were repeatedly added after 24 and 48 h. Surface expression of the inhibitory molecules PD-1 and TIM3 on effector cells was assessed by flow cytometry.
Statistical analysis
The data were expressed as the mean ± standard deviation (SD) and analyzed with software Prism software v9.0. The differences between two independent groups were compared using Student’s t test. One-way ANOVA was used when three or more independent groups were compared. P < 0.05 was set as the significant difference level.
Results
Generation of anti-CD22 CAR NK cells
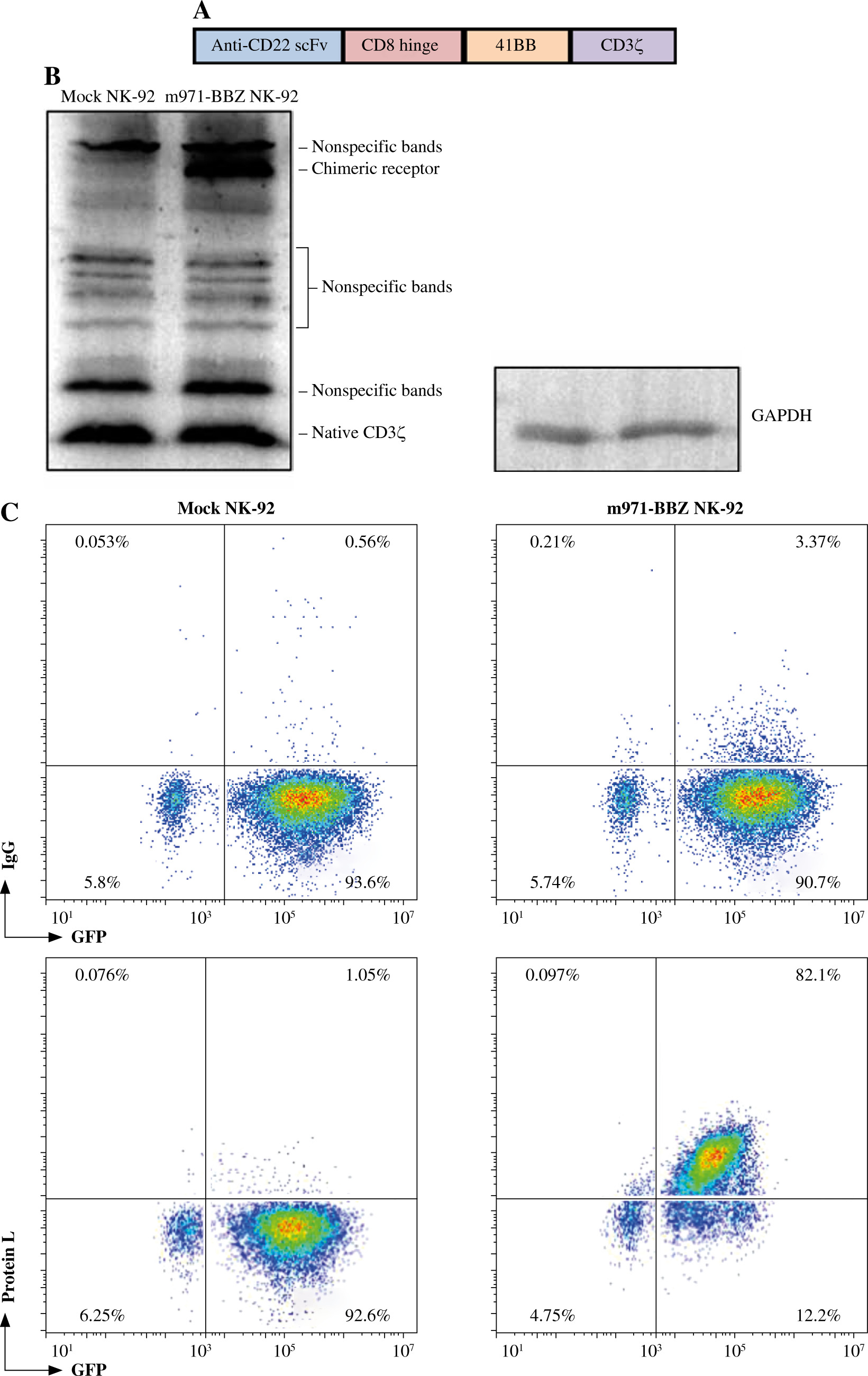
First, we designed a second-generation CD22-specific chimeric antigen receptor which consists of anti-CD22 scFv and transmembrane regions of the CD8 molecule, the 41BB costimulatory signaling moiety, and the cytoplasmic component of the CD3ζ molecule. In addition, we incorporated the EGFP reporter gene to evaluate the efficiency of transduction (Fig. 1A). To confirm whether the m971-41BB-CAR was transduced successfully, the EGFP positive NK-92 cells were sorted and expanded, and then they were subjected to western blotting with anti-CD3ζ mAb (Fig. 1B). In contrast with mock NK-92 cells, which only expressed endogenous CD3ζ protein (16 kDa), m971-BBZ NK-92 cells not only expressed native CD3ζ at the predicted site, but also obviously expressed the chimeric m971-scFv-41BB-CD3ζ fusion protein (55 kDa). Finally, we detected the expression of m971-BBZ on the surface of NK-92 cells by staining with biotin-labeled protein L, which could recognize the scFv portion of anti- CD22. The results of flow cytometry analysis revealed that 82.1% of m971-41BB-CAR was expressed on surface of NK-92 cells, while the CAR expression remained almost undetected on the mock NK-92 cells (Fig. 1C). Taken together, the observations indicate that the CD22-specific chimeric antigen receptor was expressed on the surface of m971-BBZ NK-92 cells successfully.
Fig. 1
Generation of m971-specific CAR NK-92 cells. A) Schematic representation of m971-specific CAR lentiviral vector. B) Immunoblotting analysis of CAR expression. The cell lysates of mock or m971-CAR infected NK-92 cells were subjected to Western blot analysis with anti-human CD3ζ primary antibody. C) Flow cytometric analysis of CAR expression. Mock or m971-BBZ NK-92 cells were stained with biotin-labeled protein L or isotype-matched control antibody, followed by APC-labeled anti-streptavidin antibody staining to define the expression of CAR
In vitro cytolytic potency against lymphoma cell lines triggered by CD22-specific CAR
We detected the expression of CD22 on the surface of the three common lymphoma cell lines Daudi, Raji and TMD8 by flow cytometry. Obviously, the three lymphoma cell lines all highly expressed CD22 protein (Fig. 2A). To define the capacity of m971-BBZ NK-92 cells for recognition of lymphoma cells that express endogenous CD22, we measured the secretion of IFN-γ and TNF-α by the method of ELISA (Fig. 2B). In contrast with mock NK-92 cells, m971-BBZ NK-92 cells stimulated with lymphoma cell lines secreted greater amounts of IFN-γ and TNF-α. The results suggest that m971-BBZ NK-92 cells can more specifically recognize lymphoma cell lines with high levels of endogenous CD22 than mock NK-92 cells.
Subsequently, to define whether the m971-BBZ NK-92 cells can lead to more efficient lymphoma cell line lysis, a bioluminescence assay was performed. Mock NK-92 cells and m971-BBZ NK-92 cells were co-cultured with three lymphoma cell lines, which express high levels of CD22 and firefly luciferase. After 6 hours the bioluminescence values were detected. We found that m971-BBZ NK-92 cells can trigger higher specific killing than mock NK-92 cells at all E : T ratios tested (Fig. 2C). Next, we further measured degranulation of NK-92 cells by detecting the expression of CD107a in mock NK-92 cells and m971-BBZ NK-92 cells incubated with lymphoma cell lines. In agreement with the cytotoxicity result, m971-BBZ NK-92 cells displayed higher CD107a expression than NK-92 cells in the presence of CD22 positive lymphoma cells, suggestive of augmented degranulation by virtue of CAR expression (Fig. 2D, E). In line with that, m971-BBZ NK-92 cells could not induce CD22-independent degranulation against 293 T cells which were CD22 negative (Fig. 3).
Fig. 2
Specific cytotoxicity and cytokine production of m971-BBZ NK-92 cells against lymphoma cell lines. A) Flow cytometric analysis of CD22 expression on the surface of lymphoma cell lines. The three lymphoma cell lines indicated were stained with APC-conjugated anti-CD22 mAb antibody (solid line) or isotype-matched control antibody (dotted line). B) Secretion of cytokines by CAR NK-92 cells activated by CD22-expressing lymphoma cells. Mock or m971-BBZ NK-92 cells were co-cultured with lymphoma cells or cultured alone (no target) for 24 hours, NK cells kept without target cells were included as controls. The supernatants were collected to measure IFN-γ and TNF-α secretion using the method of ELISA. Mean values ±SD are shown; n = 3; ***p < 0.001, **p < 0.01, *p < 0.05, ns, p ≥ 0.05. C) Cytotoxic activity of CAR NK-92 cells against lymphoma cell lines. Three lymphoma cell lines infected with firefly luciferase co-cultured with mock NK-92 cells or m971-BBZ NK-92 cells respectively for 6 hours. The cytotoxicity was determined as the bioluminescence values which were measured with substrate of firefly luciferase. Mean values ± SD are shown; n = 3, ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, ns, p ≥ 0.05.

Fig. 2
Cont. D) Degranulation of CAR NK-92 cells upon activation by CD22-positive lymphoma cells. Mock or m971-BBZ NK-92 cells were stimulated with the same number of 3 lymphoma cell lines respectively, and flow cytometric analysis of CD107a (a marker of degranulation) expression on the surface of efficient cells (mock or m971-BBZ NK-92 cells) was performed. NK cells kept without target cells were included as controls. Data shown are representative for three independent experiments
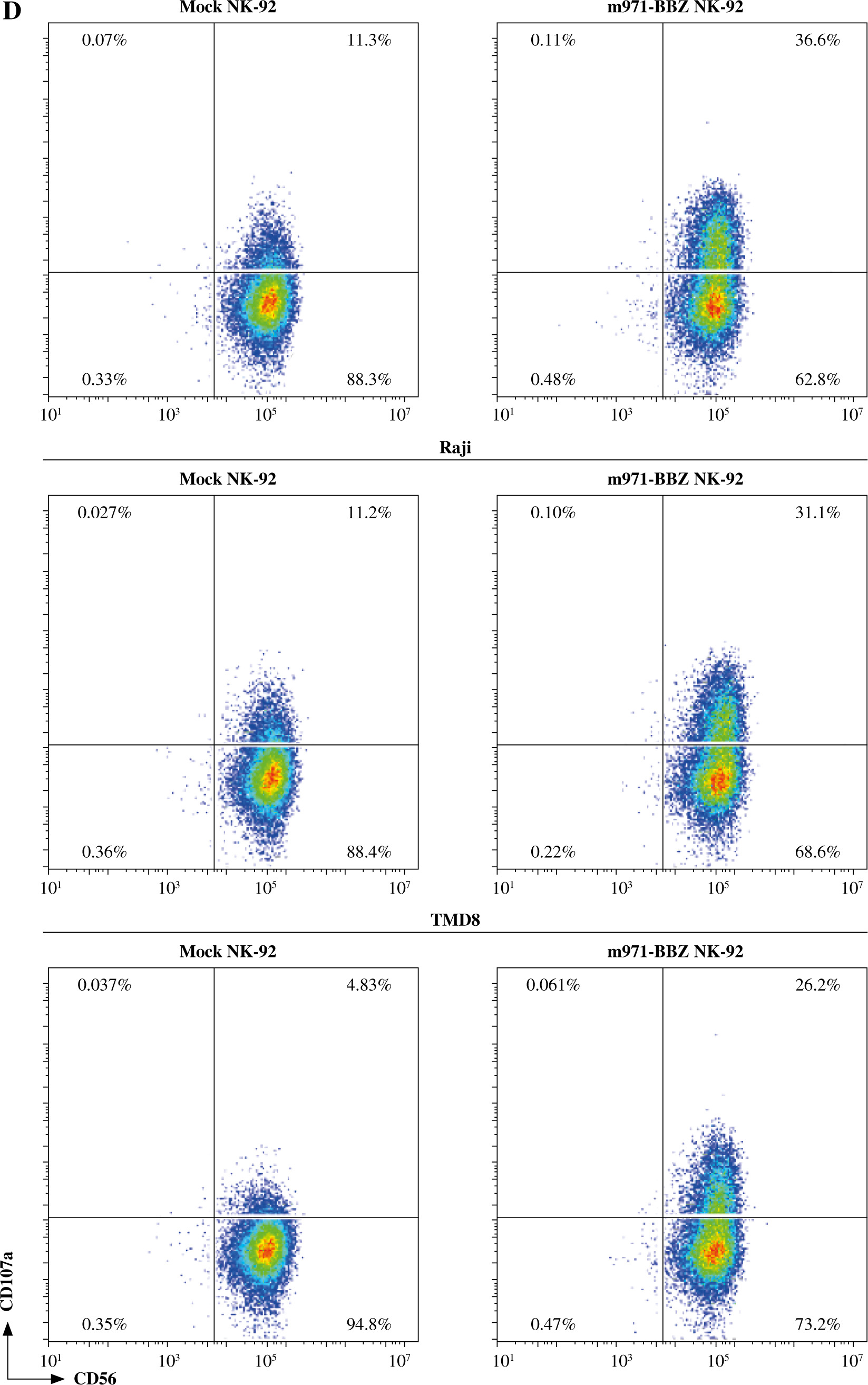
Fig. 2
Cont. E) Statistical analysis of expression level of CD107a of activated CAR NK-92 cells. Mean values ±SD are shown; n = 3; ****p < 0.0001, *p < 0.05
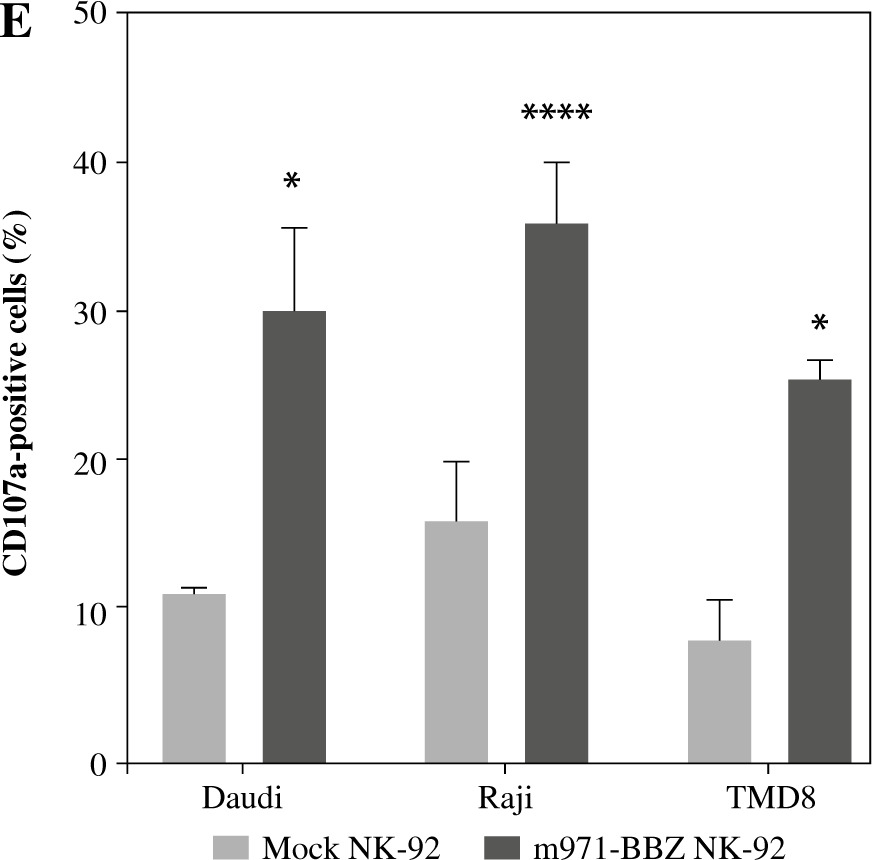
Fig. 3
Degranulation of m971-BBZ NK-92 cells against 293T cells. A) Flow cytometric analysis of CD22 expression on the surface of 293T cells. B) Degranulation of CAR NK-92 cells against 293T cells. Flow cytometric analysis of CD107a expression on the surface of effector cells (mock or m971-BBZ NK-92 cells) was performed after being cocultured with 293T cells for 4 hours. Data shown are representative for three independent experiments. C) Statistical analysis of expression level of CD107a of CAR NK-92 cells against 293T cells. Mean values ±SD are shown; n = 3; ns, p ≥ 0.05
Cytotoxic activity of CD22-CAR NK-92 cells against primary lymphoma cells
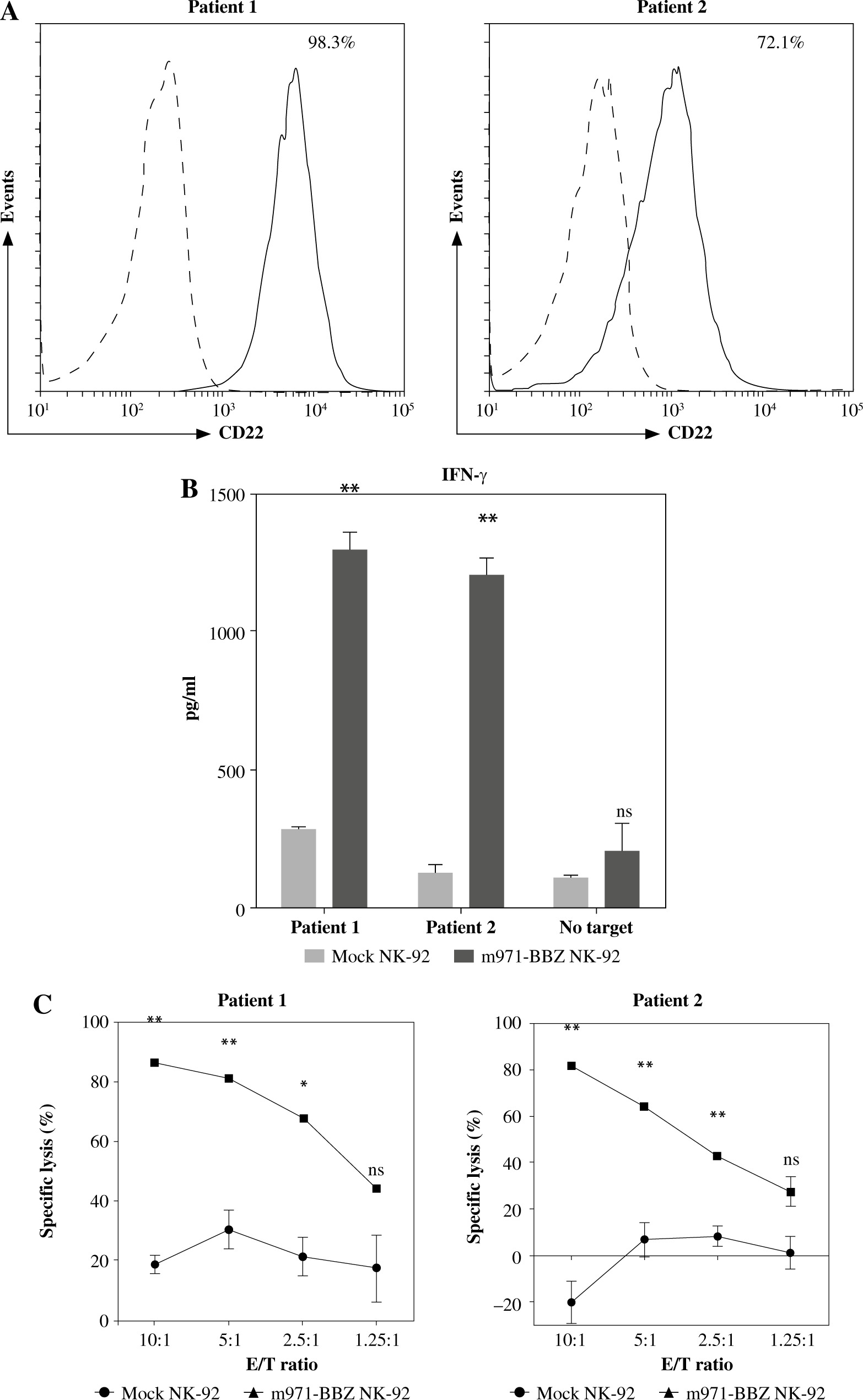
We harvested lymph node biopsies from two patients with B cell lymphoma and detected the expression of CD22 in lymphoma cells by flow cytometry analysis, and found that CD22 was highly expressed in primary lymphoma cells from both patients (Fig. 4A). Then, to define the capacity of m971-BBZ NK-92 cells for recognition of CD22+ cells isolated from the patients with B cell lymphoma, we measured the secretion of IFN-γ by the method of ELISA (Fig. 4B), with a higher level of IFN-γ detected in the m971-BBZ NK-92 group. Moreover, we measured the cytotoxic activity of anti-CD22 CAR NK-92 cells against those primary lymphoma cells, as shown in Fig. 4C. m971-BBZ NK-92 cells displayed more potent cell killing activity at the E : T ratios of 10 : 1, 5 : 1 and 2.5 : 1. The results revealed that m971-BBZ NK-92 cells could efficiently recognize and lyse primary lymphoma cells.
Fig. 4
m971-BBZ NK-92 cells co-cultured with CD19+ cells isolated from patients with B cell lymphoma respectively achieved remarkably higher cytotoxic activity than mock NK-92 cells. A) Flow cytometric analysis of CD22 expression on the surface of CD19+ cells from patients with B cell lymphoma. The CD19+ cells were stained with APC-conjugated anti-CD22 mAb antibody (solid line) or isotype-matched control antibody (dotted line). B) Secretion of cytokines by CAR NK-92 cells activated by primary lymphoma cells. CD19+ cells derived from two patients were co-cultured with mock NK-92 cells or m971-CAR NK-92 cells, and the secretion of IFN-γ was measured by ELISA. Mean values ±SD are shown; **p < 0.01, ns, p ≥ 0.05. C) Cytotoxic activity of CAR NK-92 cells against primary lymphoma cell lines. CD19+ cells derived from two patients were stained with eFluor 670 and co-cultured with mock NK-92 cells or m971-BBZ NK-92 cells for 6 hours, and then the specific lysis was detected with the method of absolute counting. Mean values ±SD are shown, **p < 0.01, *p < 0.05, ns, p ≥ 0.05
The impact of irradiation on the cytolytic effect and repeated stimulation on the persistence of CD22-CAR NK-92 cells
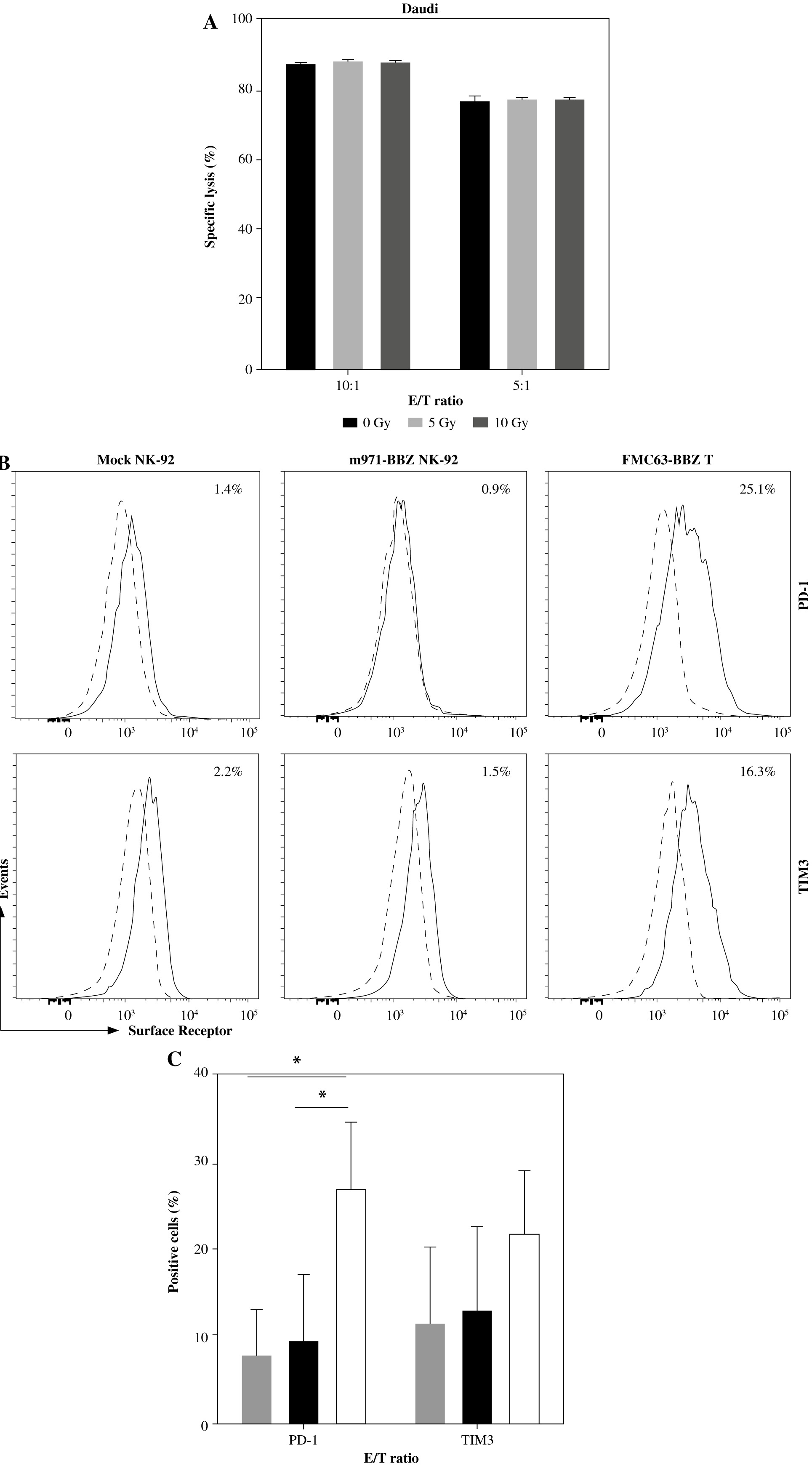
To determine whether different dosages of X-rays can influence the cytolytic potency of m971-BBZ NK-92 cells, we treated m971-BBZ NK-92 cells with two different dosages of X-rays, and then m971-BBZ cells were co-cultured with Daudi cells. After 6 hours, we detected the specific killing and found that the specific killing efficiency of m971-BBZ NK-92 cells was not influenced by the two different irradiation dosages (Fig. 5A). In addition, we assessed the effect of repeated stimulation on CAR- and mock-transduced NK-92. Here, we included the commonly used CD19-targeting FMC63-BBZ T cells as the control, which have been reported in our previous study [22]. After repeated stimulation with TMD8 cells, the surface expression of inhibitory receptors PD-1 on m971-BBZ NK-92 cells exhibited a lower level than that on FMC63-BBZ T cells (Fig. 5B), while the expression level of TIM3 was increased both in CAR NK-92 and FMC63-BBZ T groups (Fig. 5C).
Fig. 5
Effects of irradiation on the cytotoxicity and antigen repeated stimulation on the persistence of CAR NK-92 cells. A) Cytotoxic activity of CAR NK-92 cells after irradiation. m971-BBZ NK-92 cells were irradiated with different doses of X-rays and co-cultured with Daudi cells for 6 hours which were infected with firefly luciferase, and the cytotoxicity was measured. B) Effect of repeated stimulation on CAR NK-92 cells. Inhibitory surface receptor expression of PD-1 and TIM3 on m971-BBZ, mock-transduced NK-92 cells and FMC63 BBZ CAR-T cells repeatedly stimulated (solid lines) was analyzed. Cells stained with isotype antibody served as controls (dashed lines). Data shown are representative for three independent experiments. C) Statistical analysis of inhibitory receptor expression of PD-1 and TIM3 on effector cells. Mean values ±SD are shown; n = 3; *p < 0.05
Discussion
Compared to T cells, NK cells provide a CAR-engineering platform that is safer and more advantageous [23]. As a source of NK cells, NK-92 cells can be manipulated more easily to express receptors or ligands than peripheral blood NK cells and demonstrated consistent cytotoxic activity against cancer targets [10]. In this study, we constructed a second generation CD22-specific CAR harboring scFv antibody fragment and CD8α hinge region, linked to human composite 41BB (CD137)-CD3ζ signaling domains. To explain our choice of CAR construction, a study proved that the second-generation CAR-NK constructs displayed increased cytotoxicity compared to the first generation [24]. The costimulatory protein domains exhibited different properties: while CD28-CD3ζ CARs led to stronger T-cell activation, 41BB-CD3ζ CARs prolonged in vivo T-cell persistence and reduced exhaustion, which seemed to be more suitable for clinical application [25, 26].
CD22-directed CAR-NK-92 cells (m971-BBZ NK-92) obtained via lentiviral gene transfer displayed homogeneous and stable CAR surface expression, and showed high and specific cytotoxicity against CD22-expressing lymphoma cell lines and primary patient cells even at low E/T. Compared with mock NK-92 cells, m971-BBZ NK-92 cells produced a significantly increased amount of IFN-γ and TNF-α, which were typical for activated NK cells and supported the CAR NK-92-induced activation of endogenous antitumor immunity. Consistent with the data regarding cytolytic activity and cytokine release, a greater extent of degranulation was observed in m971-BBZ NK-92 cells responding to CD22-positive target cells than mock NK-92 cells. The CD22-dependent degranulation of m971-BBZ NK-92 cells was further supported by the evidence that 293 T cells negative for CD22 expression only triggered negligible NK-cell degranulation. More importantly, in a more clinically relevant context, we demonstrated that m971-BBZ NK-92 cells efficiently recognized and eradicated primary lymphoma cells derived from two patients ex vivo. It should be admitted here that the fact that the experiment was performed on only two patient biopsies was the limitation of this study, although the effect of CAR-NK92 cells was really impressive. Even though we have previously tried our best to obtain more patient samples, unfortunately only two patient samples have been available for our analysis so far for many reasons, and we would like to expand the research group to consolidate this conclusion in the future.
As the NK-92 cells which used in the clinical trials are aneuploid, they must be irradiated before being administered to patients [10]. In phase I clinical trials irradiation of untargeted NK-92 cells with 10 Gy prior to infusion had been included as a safety measure to prevent permanent engraftment [7]. This raises the concern that irradiation may dampen the cytotoxicity of m971-BBZ NK-92 cells. Fortunately, based on our data, irradiation at the tested dosages (5 Gy and 10 Gy) did not apparently impair the cytotoxicity of m971-BBZ NK-92 cells. Meanwhile, irradiated NK-92 cells have a limited lifespan in vivo and do not develop memory, minimizing the risk of these side effects [27].
Persistence of the CAR-expressing cells is another aspect for further clinical application. The elevated surface expression of inhibitory molecules such as PD-1 and TIM3 has been associated with exhaustion in CAR-expressing NK cells and other lymphocytes [28]. The flow cytometry results indicated that repeated antigen stimulation with CD22-positive target cells affected the expression of PD-1 and TIM3 differently in our CAR-NK92 cells and CAR-T cells. Compared to FMC-BBZ T cells, repeated antigen stimulation had no major effect on expression of PD-1. Importantly, a recent study revealed that PD-1 expression level remained low after continuous activation of CAR-NK-92 cells with 41BB-CD3ζ signaling domains [29], which was in line with our results of the detection of the inhibitory receptors and indicated an advantage over CAR-T therapy that can be inhibited by PD-L1 from target cells. Conversely, TIM3 expression increased in both m971-BBZ NK-92 and FMC63-BBZ T cells following repeated stimulation with target cells. Nowadays, one of the limitations of CAR-T therapies applied in chronic lymphocytic leukemia and solid tumors is inhibitory activity or a state of exhaustion of T cells [30]. The CAR-expressing cells exposed to chronic antigen stimulation tend to become exhausted, as characterized by impaired expansion, increased expression of inhibitory receptors and lower cytokine production [31]. It is of great importance to uncover the molecular mechanism affecting the persistence of CAR-expressing cells in further research, which could augment the efficiency of CAR-based immunotherapy.
Recently, abundant clinical data have demonstrated that existing targets for CAR engineering cannot meet the requirements, as cunning tumor cells acquired the ability to lose or diminish surface expression of the specific antigen. Our team has also made great effort to explore alternative targets for cancer retargeting in previous research [22, 32]. This study demonstrated that CD22 is a promising antigen for targeting by CAR-NK-92 cells in the treatment of B cell lymphoma. This may contribute to developing CAR-NK immunotherapy toward clinical applications.