
Introduction
Diseases of the lower extremities are often associated with chronic venous disease, which can lead to varicose vein disease. Varicose vein diseases are characterized by veins that twist, dilate, and bulge [1]. Because they facilitate one-way inward blood flow through competent valves and link the superficial and deep venous systems, perforator veins are essential to the venous circulation of the lower limbs. The calf muscle pump, which generates pressure gradients that force blood toward the heart, is the main force behind this transfer. The great saphenous vein, perforator veins, small saphenous vein, and other subcutaneous veins in the leg can all malfunction due to valve incompetence. Venous reflux and increased venous pressure brought on by such incompetence can cause vein dilatation (diameter ≥ 3 mm) and other clinical symptoms such as edema, skin hyperpigmentation, and, in more severe cases, venous ulcers [2–4]. The disease is associated with risk factors that include increased age, hormonal changes, pregnancy, excess weight, and prolonged standing [5]. Varicose veins become increasingly prevalent as people age, most likely due to weakened calf muscles and a decrease in the matrix components of the veins. The occurrence of the three varicosity classes rises linearly with aging [6]. Women are more likely than men to have varicose veins, and the severity increases with each additional pregnancy, according to research linking the disease to gender. An increase in blood volume, intra-abdominal pressure, the release of the potent vasodilator relaxin from progesterone production, and an increase in venous capacitance caused by estrogen are some of the factors contributing to venous relaxation. According to research, women who are overweight or obese are more likely to develop varicose veins. A similar relationship does not appear to exist for men [7, 8]. According to recent research, a disruption in the regulatory vessel wall homeostasis mechanisms is a possible factor in the development of varicose veins. Alterations in angiogenesis may be a major contributing factor to the pathophysiology of this illness. The process of creating new blood vessels is called angiogenesis. It is a multi-step process in both healthy and diseased circumstances, controlled by endogenous regulators, both positive and negative [7], and various types of cellular receptors and binding proteins in blood vessel walls, resulting in modifications to the lumen. Through intracellular communication, these receptors/proteins can transfer information to the blood vessel tube’s interior, vascular tissue, and the surrounding supporting structures. Modifications as blood passes through the vein cause blood pressure to distort the wall. The vein and the intracellular signaling pathway may be altered due to this pressure [9]. One possible explanation for these contradictory results is that the notion of varicose disease arising from an ineffective valve or an aberrant vein wall structure is not merely theoretical – it is the first stage. In the early stages, collagen biosynthesis (or decreased catabolism) can partially compensate for elastin loss without replacing the vein’s primary structure; in the end, the vein dilates, elastin and collagen levels significantly decline, and elasticity and strength are lost [10] Therefore, elastin metabolism requires a balance between elastases and elastase inhibitors. Consequently, the relative deficiency of elastin in the varicose vein wall may result from excessive elastin degradation, impaired anti-elastase activity, or a combination of both. In our study, the two major elastase inhibitors, a1-antitrypsin and a2-macroglobulin, were not systemically defective in any of the patients [11]. Often, protein in normal venous blood seen in the lower extremities is a balance between elastin, collagen, and proteoglycan contents of the extracellular matrix that provide elasticity and distensibility of the veins [12]. On the other hand, elastin was one of the most important components of vascular extracellular structures, and regular, decreased, and/or structural changes were displayed in varicose vein walls [13]. Therefore, with noticeable alterations in collagen and elastin tissue content, varicose veins also exhibit an imbalance between the primary constituents of ECM proteins. As a result, an imbalance in collagen will prevent the varicose veins from stretching. The loss of structural proteins (elastin and type 3 collagen) from the vessel wall results in the loss of the biophysical properties of the varicose vein wall [14]. Also, compounds that mediate dilatation are added to elastin. Blood vessels are regulated by endothelial growth factor (VEGF), which plays a central role in preserving the integrity and reactivity of the blood vessels. VEGF mediates dilatory responses and vaso-permeability in conjunction with nitric oxide (NO) [15]. Most research has focused on vascular endothelial growth factor A (VEGF-A) due to its critical role in regulating angiogenesis. The VEGF receptors regulate the subsequent intracellular signals in conjunction with other members of the VEGF family. The activation of downstream pathways is facilitated by dimeric VEGF binding to the extracellular domains of VEGFR, which promotes receptor dimerization and mutual tyrosine transphosphorylation of the intracellular domains between receptor subunits. VEGF ligands specifically bind to three different cell membrane-bound receptors: VEGFR1 (Flt-1), VEGFR2 (Flk/KDR), and VEGFR3 (Flt-4) [16]. According to previous research, VEGF interacts with its receptors to facilitate its biological functions by inducing BAX and BCL-2. But the main receptor that mediates the most well-known cellular reactions to VEGF is VEGFR2 [17]. A key player in maintaining vascular reactivity and integrity is VEGF-A, a regulator of angiogenesis in healthy and diseased conditions. It is a selective endothelial cell mitogen that stimulates endothelial cell migration, proliferation, and differentiation by binding to VEGFR2 or VEGFR1 receptors. While VEGFR1 is merely a decoy receptor, VEGF2 is the primary signaling receptor of VEGF-A, regulating almost all of its biological effects in endothelial cells. VEGF-A promotes endothelial nitric oxide synthase expression and synthesis [18, 19], causing a rise in venous pressure in the lower limb veins as a result of extended standing. This results in mechanical stimulation of endothelial cells and vascular smooth muscle as well as peripheral tension on the artery wall, where intracellular signaling toward vascular function modification is regarded as an alarm. These changes may occur directly or indirectly, influencing factors such as vascular aging. Activation can arise through many methods, such as elevated reactive oxygen species (ROS) and oxidation levels in cells [20], reduced NO levels, stiffened vascular walls, and gene expression [21–24]. However, the idea that valvular incompetence promotes hydrostatic phenomena that cause varicosities leads to the hypothesis that vascular tone plays a major role in the pathophysiology of lower extremity varicose veins. In addition to controlling vascular tone, NO, a strong intercellular molecular messenger, also has a variety of pathophysiologic roles, including cytotoxicity, neurotransmission, mediation of the inflammatory cascade, and inhibition of platelet adhesion/aggregation [25]. Despite varicose vein disease being widespread, little is known about the molecular processes underlying venous wall remodeling. A dearth of thorough information combining angiogenic signals, vascular tone regulators, and structural proteins such as elastin exists despite earlier research having separately examined elements like VEGF or NO. Additionally, little research has looked at the relationship between these biomarkers and the severity of the disease, especially in populations that are underrepresented. By concurrently assessing VEGF, VEGFR, NO, and elastin in patients with varicose veins and investigating their clinical implications, this study aimed to close these gaps.
Material and methods
This study was prospective and nonrandomized. There were 60 patients with lower extremity primary varicose veins and 60 patients with severe varicose vein diseases between January and March 2025. A control group of 60 healthy adults was employed.
Inclusion criteria
Women with primary and severe varicose veins in their lower extremities were included in the study. A total of 120 patients were enrolled following the application of the inclusion and exclusion criteria. Patients were grouped according to the American Venous Forum’s CEAP classification: clinical (C), etiological (E), anatomical (A), and pathophysiological (P).
The mild group, which corresponds to CEAP class C2, had blue varicose veins that were visible and had a limited diameter without causing noticeable limb changes. In contrast, the severe group showed extensive segmental involvement and noticeably dilated, tortuous varicose veins, which is consistent with advanced CEAP classification (C3–C4) [26]. Patients with atherosclerosis, cancer, arteriovenous malformations, chronic illnesses, or coexisting acute diseases were not included in this study; they were between the ages of 30 and 50. Medication was not administered to patients.
Control group
Sixty healthy women were chosen to form the control group. They did not have lower extremity varicose veins or chronic venous insufficiency, and they were not on any long-term medications or had taken any in the previous 2 weeks. The control group’s members were selected based on their venous symptoms, history of venous disease, or absence of visible varicose veins. A clinical examination showed that the limbs appeared normal and that there were no signs of venous insufficiency.
Collection and management of samples
Blood samples were extracted from the brachial vein. The samples were collected by drawing blood into serum tubes through venipuncture. The materials were subsequently centrifuged for 7 to 10 minutes at 4000 rpm. The supernatant was transferred to several Eppendorf tubes and stored in freezers at –80°C.
Assessment of serum growth factor concentrations
Levels of serum growth factors were assessed using the enzyme-linked immunosorbent sandwich assay (ELISA) method according to the manufacturer’s specifications. Human ELISA kits for VEGF, vascular endothelial growth factor receptor (VEGFR), NO, and elastin were employed.
This study utilized BT-LAB (UK). Samples and prepared standards were placed into 96-well plates and incubated at room temperature for 2 hours. Following buffer washing, the premade biotin-conjugate was added and allowed to sit at room temperature for 2 hours. After that, premade streptavidin-HRP was added, and the mixture was incubated for 1 hour at room temperature. Then, TMB was added. Substrate solution was kept at room temperature for 30 minutes, avoiding exposure to direct sunlight. The stop solution that was supplied then brought the reaction to an end. Plates were promptly measured with a microwell ELISA reader spectrophotometer with the primary wavelength set at 450 nm. Then the absorbance was measured. Mathematical formulae were applied to the readings to compute sample results and create a standard curve.
Statistical evaluations
Data are presented as mean ± standard deviation. The statistical analyses were conducted using SPSS software [27, 28]. Nonparametric tests without pairing were used. A one-way ANOVA was employed to compare the means [29, 30]. Statistical significance was established at p < 0.05. Furthermore, correlations among the parameters were evaluated [31, 32].
Results
Descriptive analysis of biomarkers
The results of comparing patients with mild and severe varicose veins with healthy controls are shown in Table I. Significant biochemical changes were observed. Interestingly, levels of VEGF-A were considerably increased in both the mild and severe varicose veins groups compared to the control group (p < 0.05).
Table I
Level of measured parameters in mild cases, severe cases, and controls
[i] Data are presented as mean ± SD: standard deviation, p > 0.05 – significant differences, BMI – body mass index, CRP – C-reactive protein, ESR – erythrocyte sedimentation rate, VEGF – vascular endothelial growth factor, VEGFR – vascular endothelial growth factor receptor, ELN – elastin, NO – nitric oxide, p1 – the comparison between mild and severe groups, p2 – the comparison between mild and control groups, p3 – the comparison between severe and control groups.
Similarly, levels of VEGFR2, a vascular endothelial growth factor receptor, were significantly higher in both patient groups compared to the control group (p < 0.05).
In addition, patients with mild and severe varicose veins had significantly higher NO concentrations than healthy controls, indicating endothelial dysfunction (p < 0.05). Furthermore, patients with varicose veins had significantly higher elastin levels, and the increase was more pronounced in those with more severe disease (p < 0.05).
However, no statistically significant differences were observed in zinc (Zn) levels between the two groups. However, both groups of patients showed significantly higher levels of inflammatory biomarkers compared to the control group, including erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) (p < 0.05). Furthermore, the severity of varicose veins was found to be closely associated with body mass index (BMI) (p < 0.05).
Correlation analysis
Table II shows the correlation analysis between the studied biomarkers. The results show that the correlations were both negative and positive, indicating an inverse and direct relationship between the biomarkers. More specifically, vascular endothelial growth factor (VEGF-A) showed a statistically significant weak to moderate positive correlation with BMI (r = 0.325**, p = 0.002), CRP (r = 0.397**, p < 0.001), and ESR (0.396**, p < 0.001), indicating that higher BMI, CRP, and ESR levels are associated with higher VEGF levels. While the correlation was very strongly positive for VEGF-A receptor type II (r = 0.774**, p < 0.001), it was moderately positive for elastin (ELN) (r = 0.394**, p < 0.001), and moderately to strongly positive for NO (r = 0.576**, p < 0.001). This suggests that their increase is associated with increased VEGF-A. However, lower positive correlations were observed with zinc (r = 0.132, p = 0.215). It also showed a weak to moderate positive correlation indicating a direct relationship between VEGFR2 and BMI (r = 0.322**, p = 0.002), CRP (r = 0.439**, p < 0.001) and ESR (r = 0.407**, p < 0.001) and ELN (r = 0.442**, p < 0.001), while indicating a very strong positive correlation with NO (r = 0.682**, p < 0.001). However, no correlation was found with Zn (r = –0.028, p = 0.790). Furthermore, ELN showed a significant direct relationship with BMI (r = 0.447**, p < 0.001), CRP (r = 0.590**, p < 0.001), ESR (r = 0.575**, p < 0.001) and NO (r = 0.293**, p = 0.005), which showed a lower to moderate correlation, yet did not correlate with Zn (r = –0.073, p = 0.497). On the other hand, NO had a weak to moderate positive correlation with BMI (r = 0.310**, p = 0.003), CRP (r = 0.305**, p = 0.003), and ESR (r = 0.258*, p = 0.014), while Zn (r = 0.062, p = 0.564) showed no correlation. These results suggest that proangiogenic factors (VEGF-A, VEGFR2, ELN, NO) tend to increase, highlighting potential pathophysiological interactions in varicose vein disease.
Table II
Correlations between measured parameters in varicose vein patients
| BMI | CRP [mg/l] | Zn [μg/day] | ESR [mm/h] | ELN [μg/ml] | VEGF-A [ng/l] | VEGFR2 [ng/ml] | NO [μmol/l] | |
|---|---|---|---|---|---|---|---|---|
| BMI | ||||||||
| Pearson correlation | 1 | 0.422** | –0.044 | 0.388** | 0.447** | 0.325** | 0.322** | 0.310** |
| Sig. (2-tailed) | 0.000 | 0.682 | 0.000 | 0.000 | 0.002 | 0.002 | 0.003 | |
| CRP [mg/l] | ||||||||
| Pearson correlation | 0.422** | 1 | –0.213* | 0.617** | 0.590** | 0.397** | 0.439** | 0.305** |
| Sig. (2-tailed) | 0.000 | 0.044 | 0.000 | 0.000 | 0.000 | 0.000 | 0.003 | |
| Zn [μg/day] | ||||||||
| Pearson Correlation | –0.044 | –0.213* | 1 | 0.016 | –0.073 | 0.132 | –0.028 | 0.062 |
| Sig. (2-tailed) | 0.682 | 0.044 | 0.880 | 0.497 | 0.215 | 0.790 | 0.564 | |
| ESR [mm/h] | ||||||||
| Pearson correlation | 0.388** | 0.617** | 0.016 | 1 | 0.575** | 0.396** | 0.407** | 0.258* |
| Sig. (2-tailed) | 0.000 | 0.000 | 0.880 | 0.000 | 0.000 | 0.000 | 0.014 | |
| ELN [μg/ml] | ||||||||
| Pearson correlation | 0.447** | 0.590** | –0.073 | 0.575** | 1 | 0.394** | 0.442** | 0.293** |
| Sig. (2-tailed) | 0.000 | 0.000 | 0.497 | 0.000 | 0.000 | 0.000 | 0.005 | |
| VEGF-A [ng/l] | ||||||||
| Pearson correlation | 0.325** | 0.397** | 0.132 | 0.396** | 0.394** | 1 | 0.774** | 0.576** |
| Sig. (2-tailed) | 0.002 | 0.000 | 0.215 | 0.000 | 0.000 | 0.000 | 0.000 | |
| VEGFR2 [ng/ml] | ||||||||
| Pearson correlation | 0.322** | 0.439** | –0.028 | 0.407** | 0.442** | 0.774** | 1 | 0.682** |
| Sig. (2-tailed) | 0.002 | 0.000 | 0.790 | 0.000 | 0.000 | 0.000 | 0.000 | |
| NO [μmol/l] | ||||||||
| Pearson correlation | 0.310** | 0.305** | 0.062 | 0.258* | 0.293** | 0.576** | 0.682** | 1 |
| Sig. (2-tailed) | 0.003 | 0.003 | 0.564 | 0.014 | 0.005 | 0.000 | 0.000 | |
Evaluation of biomarkers using ROC curve analysis
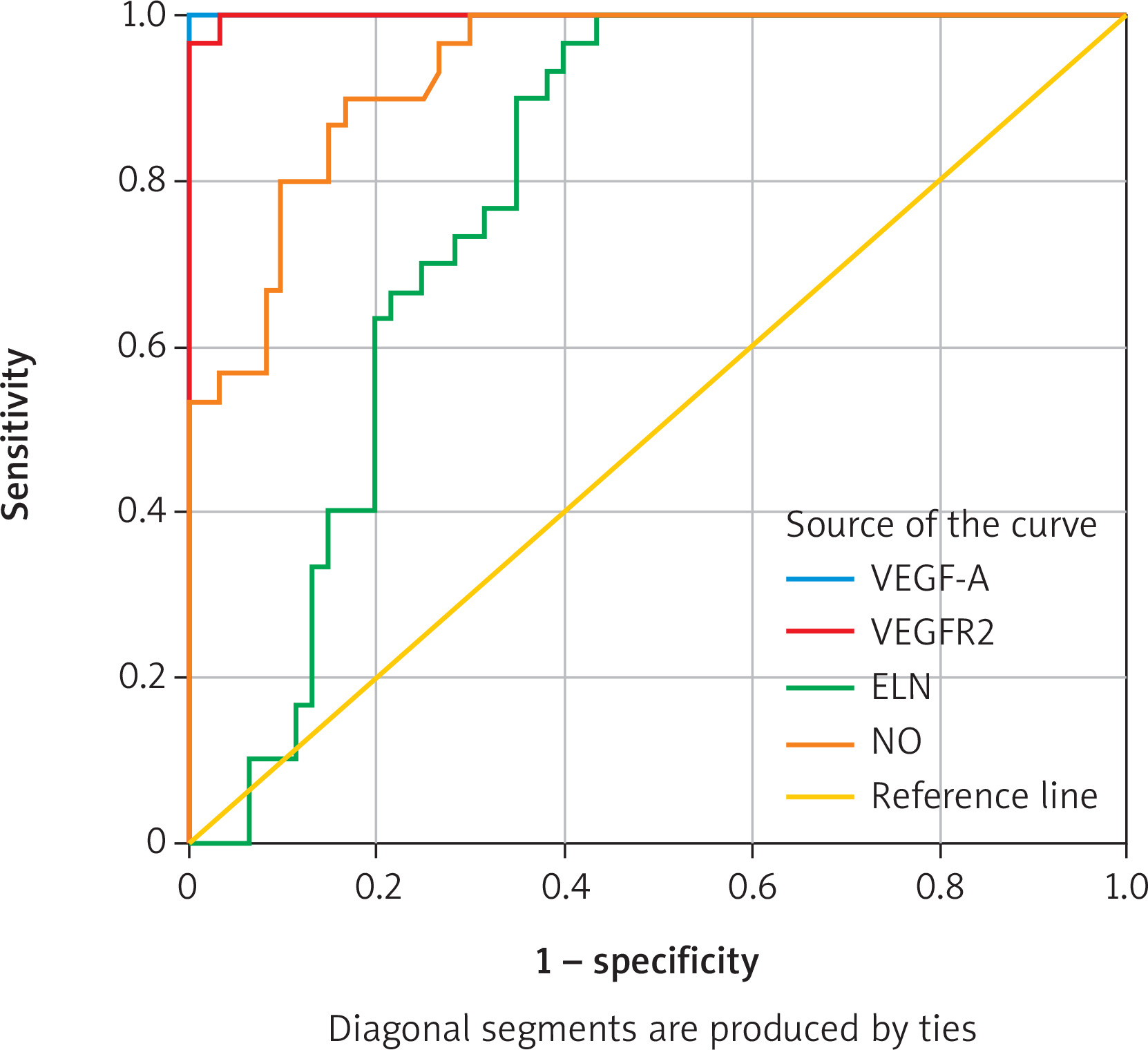
This study employed ROC curve analysis, which evaluates the accuracy of biomarkers and illustrates the relationship between sensitivity and specificity at various values. Diagnostic skill increases with the area under the curve (AUC). This analysis also enhances the clinical value of each marker, facilitating the determination of the optimal cut-off value. For this purpose, biomarkers (VEGF-A, VEGFR2, NO, and ELS) were evaluated to distinguish between patients with varicose veins and healthy controls.
The studied biomarkers exhibited varying diagnostic abilities to differentiate between patients with severe varicose veins and the control group, as demonstrated by the results of the ROC curve analysis in Table III.
Table III
Diagnostic performance of parameters with cut-off values, specificity, and sensitivity
| Item | Cut-off | Sensitivity | Specificity | AUC | 95% CI of AUC | P-value |
|---|---|---|---|---|---|---|
| VEGF | 101.1 | 100% | 100% | 1.0 | 1.0–1.0 | 0.0001 |
| VEGFR | 1.97 | 100% | 99.7% | 0.99 | 0.99–1.0 | 0.0001 |
| ELN | 23.15 | 73.3% | 71.7% | 0.784 | 0.69–0.87 | 0.0001 |
| NO | 83.18 | 86.7% | 85% | 0.93 | 0.88–0.98 | 0.0001 |
VEGF-A and VEGFR2 demonstrated the highest accuracy. VEGF-A showed an AUC of 1 (95% confidence interval: 1.0–1.0), with a sensitivity of 100% and a specificity of 100% at a cut-off of 101.1 pg/ml. VEGFR2 showed an AUC of 1 (95% confidence interval: 0.99–1.0), with a sensitivity of 100% and a specificity of 99.7% at a cut-off of 1.97pg/ml. This demonstrates their high effectiveness in distinguishing between the two groups.
NO demonstrated acceptable accuracy. It was found to have an AUC of 0.93 (95% CI: 0.88–0.91), with a sensitivity of 83.18% and a specificity of 85% at a cut-off of 83.18 μmol/l. The AUC for elastin was 0.784 (95% CI: 0.69–0.87), with a sensitivity of 73.3% and a specificity of 71.7% at a cut-off of 23.15 ng/ml. This indicates their relatively limited diagnostic role.
These results demonstrate that VEGF-A and VEGFR2 were the most effective markers in distinguishing between patients with severe varicose veins and controls, while NO and elastin were less accurate (Figure 1).
Discussion
This analysis examined the relationship between VEGF-A, VEGFR2, nitric oxide, and elastin in patients with moderate to severe varicose veins. The results showed that these biomarkers were higher in patients with both conditions than in healthy controls. VEGF-A and VEGFR2 also demonstrated high sensitivity to disease, as indicated by the ROC curve analysis. These results suggest that elevated levels of biomarkers at the site of venous damage may be a crucial factor in disease progression.
Endothelial cells form a layer that separates the blood from the vessel wall; they play a crucial role in vascular homeostasis and in adapting vessels to environmental changes. The endothelium regulates blood flow, vascular permeability, inflammatory cell recruitment, thrombosis, and angiogenesis. Disturbance of its structure and function affects vascular homeostasis [33]. One of the main factors that influences the behavior of endothelial cells is VEGF, a powerful angiogenic factor that plays a role in tissue healing and vascular remodeling, two processes that seem to be impaired in people with varicose veins [34]. By binding to the cell surface receptors VEGFR1 and VEGFR2, two tyrosine kinases present in endothelial cells of the circulatory system, VEGF-A promotes angiogenesis. The elevated expression of VEGF-A appears to play a significant role in the pathophysiology of cardiovascular disease by increasing venous wall permeability, leading to edema, and decreasing venous wall tone, which may result in venous dilatation, blood stasis in the lower extremities, and the subsequent development of venous hypertension. Additionally, elevated VEGF-A expression may influence ECM remodeling by preventing the production of various proteolytic enzymes, including matrix metalloproteinases (MMPs) [35]. According to recent data, the expression of VEGF-A in a dynamic pattern varies according to the stage of varicose vein disease [36]. The present findings demonstrated that patients with varicose veins, particularly those in advanced stages, had significantly higher levels of VEGF-A and VEGFR2, which were indicative of an inflammatory response supported by elevated CRP and ESR as well as an effort to create new blood vessels to make up for damage to the vein wall, as evidenced by elevated elastin. These results corroborate the research of Alireza Nazari et al. (2020), which demonstrated that patients with varicose veins had higher blood levels of VEGF and VEGFR2 gene expression than healthy controls, indicating increased angiogenic activity in the affected veins, which is in line with earlier research that connected varicose vein vascular inflammation to elevated VEGF-A [37]. Chronic systemic inflammation negatively affects the local angiogenesis mechanisms in varicose veins, resulting from increased ESR and CRP and increased VEGF-A and VEGFR2. This opens the door to a new understanding of the mechanism of disease progression, as VEGF is not always elevated throughout the varicose vein and may decline during the late phase of chronic inflammation. Poredos et al.’s findings forecasted hypercoagulation and an elevated local inflammatory response in varicose vein sulci. This is most likely the result of chronically elevated venous pressure and venous flow disruption, which damages the vascular wall. The evidence indicates a shared activation route and feedback control between inflammation and hemostasis. Hemostatic equilibrium may be upset during inflammation, which results in a rise in clotting factor synthesis and a fall in anticoagulant mechanism regulation [38]. VEGF-A levels rise again compared to the mild stage of the disease as the disease progresses to more severe stages (such as C4–C6). This occurs due to worsening inflammation and ischemia (lack of oxygen), which prompts the body to release VEGF-A to repair damaged vessels by promoting the growth of new blood vessels. As part of the chronic inflammatory response, VEGF is also secreted by immune cells, including neutrophils and macrophages [39, 40]. This contrasts with a study that found no increase in VEGF levels in the same patient population. Hollingsworth et al. suggest that the inability to release VEGF when blood flow is blocked may play a crucial role in the development of primary atrial fibrillation [41]. These differences suggest possible discrepancies, indicating that the role of VEGF in varicose veins may be general and dependent on several factors, such as the degree and level of hypoxia and the response of endothelial cells [42]. The equilibrium between pro- and antiangiogenic factors can be upset, which can cause either too much or too little angiogenesis, which can exacerbate medical disorders [43]. The findings align with those of another study that explained the rise. It is suggested that the receptor VEGFR2, which has been demonstrated to be expressed more strongly in the walls of affected veins than in normal veins, may be the pathway by which increased VEGF signaling in patients with varicose veins is transmitted. Given that glycocalyx components influence VEGF-A and VEGFR2 and contribute to the transmission of shear stress, vascular inflammation, and angiogenesis, this may lend credence to their role in promoting these signals [36]. VEGF-A not only helps blood vessels grow but also makes them more permeable by activating the enzyme eNOS in endothelial cells. eNOS produces NO, which lowers the resistance of the vein wall and leads to abnormal swelling. VEGF-A is a key driver of angiogenesis, increasing the production of NO from endothelial cells. It achieves this by activating both vascular endothelial growth factor receptor-1 (VEGFR1) and vascular endothelial growth factor receptor-2 (VEGFR2). Activation of VEGFR2 increases the expression and phosphorylation of eNOS, promoting NO production. NO, in turn, relaxes blood vessels and enhances vascular permeability. NO also controls the growth of VEGFR2-mediated endothelial cells and is a key part of the process of capillary development through cGMP-dependent signaling pathways. These interactions highlight the importance of VEGF-A and NO for maintaining vascular function and regulating vascular homeostasis. This VEGF-NO axis may not function properly, which can lead to health problems such as varicose veins [44]. The study also showed that NO concentrations are significantly higher in patients with severe venous disease, including skin fibrosis and healed ulcers, compared to healthy individuals. Patients in the late stages of the disease exhibited increased NO levels in their blood, indicating a clear correlation between elevated NO levels and the severity of venous disease. The findings suggest that NO may contribute to inflammatory or reparative changes in the vein wall as various diseases progress [45]. NO production is enhanced as a result. Even though NO helps reduce venous tone, it can cause endothelial dysfunction and exacerbate vascular stasis if it becomes too high due to chronic inflammation; this, in turn, can worsen varicose vein symptoms. Consequently, the correlation between high CRP and NO levels is a significant clue to an inflammatory/oxidative process that is a factor in the development of the disease. A persistent inflammatory process affecting endothelial cells is indicated by elevated levels of CRP in the context of varicose vein disease. Research has demonstrated that endothelial cells are able to express iNOS in response to inflammatory stimuli such as prostaglandin E2 and inflammatory cytokines [46]. The increase of NO in cells may not consistently confer protection. In inflammatory conditions, elevated concentrations of NO can interact with superoxide (O2 –) to produce peroxynitrite (ONOO–), a potent oxidant. This reaction signifies a pathological transition from physiological signaling to oxidative stress. Peroxynitrite has been recognized as a significant factor in endothelial dysfunction, extracellular matrix degradation, and the progressive deterioration of venous walls in chronic venous insufficiency and venous illness [47]. In the context of the disease, blood stasis and chronic high venous pressure cause oxidative stress and increased free radicals (ROS) [48].
The buildup of elastin in varicose veins may correlate with elevated NO levels. NO, recognized for its vasodilatory and signaling properties, facilitates extracellular matrix remodeling by upregulating matrix metalloproteinases (e.g., MMP-2) and growth factors, such as VEGF. These factors collectively lead to elastin overproduction and modifications in arterial wall structure, as evidenced by recent histological analyses of varicose vein tissue [21]. Genetic and histological data corroborate increased elastin deposition in varicose veins. A 2014 mRNA analysis identified substantial upregulation of elastin transcripts (p = 0.047) in varicose saphenous veins relative to controls [49].
Zinc is crucial for numerous critical functions, including nucleic acid production, cellular proliferation, and tissue growth and repair. Zinc is also an essential cofactor, playing a key role in the activity of gelatinase enzymes such as MMP-2 and MMP-9. Low zinc concentrations in tissues have been shown in some studies to lead to a decrease in the activity of these enzymes, disrupt the balance of extracellular matrix remodeling, and contribute to the degradation of elastin and the development of varicose veins [50, 51]. Our study revealed that varicose vein tissue has lower local zinc levels than healthy vessels (n = 40), which suggests an imbalance in the local oxidant-antioxidant balance and increased oxidative stress in the vein wall. This information is consistent with the results of another study [52].
The findings of this study demonstrate that complex molecular alterations in these factors contribute to the development of varicose vein disease and provide a foundation for further research into their roles, including those of zinc, VEGF-A, VEGFR2, and other mediators in regulating vascular and inflammatory processes. Future studies should focus on therapeutic approaches targeting these molecular pathways.









