
INTRODUCTION
Synthetic glucocorticoids (GCs) are widely used and highly effective drugs with anti-inflammatory and immunosuppressive effects [1-3]. Nevertheless, GC therapies are associated with severe side effects, including psychiatric effects (depression, mania, psychosis, cognitive and memory impairment) reported in up to 60% of patients regularly taking them [4, 5]. The probability of psychiatric side effects is higher with long-term treatment or high doses. Depression tends to occur with long-term GC use, whereas mania can occur much earlier in treatment and is associated with high-doses [1, 3, 5]. The paper aims to discuss the depression associated with long-term GC therapy. Available data suggest several mechanisms of depression, including changes in the serotonergic, noradrenergic, dopaminergic, and glutamatergic systems, increased inflammation, disruption of the hypothalamic-pituitary-adrenal (HPA) axis, and decreased neurogenesis and neuroplasticity [6]. The exact mechanism of glucocorticoid- induced depression is poorly understood. It is known that GC therapy, among others, disrupts HPA axis activity, impairs dopaminergic neurons, suppresses hippocampal neurogenesis, and sensitizes the amygdala. Epigenetic factors are significant for these processes, and therefore in the presented work, these processes will be discussed.
MECHANISM OF GLUCOCORTICOIDS ACTION
GCs are steroid hormones secreted by the adrenal glands, and their synthesis is regulated by the HPA axis [3]. The secretion of GCs (cortisol in humans and corticosterone in rodents) is dependent directly on corticotropin (adrenocorticotropic hormone – ACTH), secreted from the pituitary gland, and indirectly on corticotropin-releasing factor (CRF), a hormone released from the paraventricular nuclei of the hypothalamus (PVN). In addition to mediating downstream physiological effects, cortisol also provides negative feedback to the HPA axis by inhibiting the release of CRH and ACTH and, subsequently, its secretion [3].
GCs act through two mechanisms: genomic and non-genomic [2, 7, 8]. The classical genomic mechanism of action of GCs is related to glucocorticoid receptors (GR) receptors and mineralocorticoid receptors (MR). GR receptors play an important role in response to a stress factor when the concentration of GCs is high [2, 9]. MR receptors regulate diurnal variation in GC levels (the diurnal rhythm of the HPA axis) and mediate tonic inhibition of PVN by the hippocampus [10].
In the cytoplasm of the cell, MR and GR receptors are present in an inactive form, linked to proteins including heat shock (proteins that regulate cell survival, differen tiation, and death; their expression increases during stress exposure), HSP90 and HSP70 (which prevent the receptor from penetrating from the cytoplasm to the cell nucleus), as well as to a variety of co-chaperones, including FKBP5 (also known as PPIase FKBP5) and BAG1, which mask the nuclear localisation signal [2, 7]. Once the receptor is combined with GCs, the activated steroid-receptor complex enters the cell nucleus and binds to the hormone response (GRE) DNA sequence [2, 7]. Receptors for GCs, when coupled to GRE, regulate gene expression through direct activation of transcription. Inhibition of gene transcription by GCs receptors has also been shown when combined with so-called “negative GRE” (nGRE) [2, 7]. Gene repression has also been found, independent of DNA, by direct protein interaction (“cross-talk”) with other transcription factors: nuclear factor kappa B (NFκB), AP1 factor, nuclear factor of activated T cells (NFAT), and signal transducer and activator of transcription (STAT) [2, 7].
In contrast, the mechanism of the non-genomic action of GCs is not clear [8, 11, 12]. Preclinical studies have shown that GCs can produce a rapid effect by reacting with GABAA receptor-dependent chloride channels [13], direct effects on the cell membrane [12], or by activating calcium uptake from the synaptic gap after potassium ion-induced depolarisation [8].
GLUCOCORTICOIDS-INDUCED DEPRESSION – THE ROLE OF MICRORNAS
GCs may also act through epigenetic mechanisms. Epigenetics refers to gene expression that can be modi fied by environmental or external factors, resulting inheritable characteristics that do not involve changes to the underlying DNA sequence. Thus, it is a change in phenotype without a change in genotype [14]. Epigenetic mechanisms include DNA methylation, histone modifications, nucleo-some repositioning, higher-order chromatin remodelling, non-coding RNA (microRNA), and RNA and DNA editing [7]. The GR receptor can locally demethylate DNA at GRE sites, resulting in an increased transcriptional response to dexamethasone in rat hepatoma cells [7, 15]. Emerging evidence indicates that GCs influence cell proliferation and survival, and function at least in part by regulating micro-RNA expression; microRNAs also modulate GC production by the adrenal glands and the cells’ responses to GCs [16]. MicroRNAs (miRNAs) are small single-stranded non-coding RNA molecules (containing about 22 nucleotides) that can mediate in RNA silencing and post-transcriptional regu lation of gene expression [17, 18]. MicroRNAs function via base-pairing with complementary sequences within mRNA molecules [19]. The binding of microRNAs can decrease the stability of target mRNA or inhibit its translation to downregulate mRNA expression [20]. MicroRNAs occasionally also cause histone modification and DNA methylation of promoter sites, which affects the expression of target genes [21, 22]. Both preclinical and clinical studies have demonstrated that miRNAs may contribute to the pathophysiology of depression and the actions of anti-depressant drugs [23-25]. In rodents, elevated miR-124-3p levels in the brain were associated with decreased GR levels and increased depression-like symptoms [16, 20]. Dwivedi et al. [23] were the first to show that chronic corticosterone administration to rats caused the altered expression of 26 microRNAs in the prefrontal cortex (19 were upregulated and 7 were downregulated); miR-124 was the most significantly upregulated. Chronic social defeat stress increased miR-124 in the hippocampus, and miR-124a overexpression exacerbated depression-like behaviour. Moreover, selective lentivirus-mediated suppression of miR-124a (miR-124a- silencers) in the hippocampus produces antidepressant-like effects in rats [26]. A recent study has indicated that the expression of miR-124-3p is significantly upregulated in the serum of major depressive disorder patients compared with healthy control subjects, which was similar to the results of studies in corticosterone-treated rats and human post-mortem tissue [27]. MicroRNAs also play an essential role in the efficacy of GC therapy. For example, patients with acute lymphoblastic leukemia responding poorly to treatment have been shown to have high miR-124-3p expression accompanied by increased GR receptor expression [16, 28]. Additionally, the literature suggests possible roles of several other micro RNAs (e.g., miR-9, miR-34c, miR-326, miR-494, and miR-504) in processes related to stress and changes in dopaminergic system activity [29-31]. These findings suggest that specific miRNAs may be both a biomarker and therapeutic target for depression related to GC therapy.
GLUCOCORTICOIDS-INDUCED DEPRESSION – THE ROLE OF DOPAMINERGIC SYSTEM
Long-term GC therapy often leads to depression [5]. Chronic GC administration can induce inducing a decrease in GR receptor sensitivity in the medial prefrontal cortex (PFC) and/or hippocampus and a weakening of the GCs feedback mechanism, which can lead to a decrease in DA release, and consequently induces anhedonia symptoms [32, 33]. Anhedonia (a core symptom of depression) is predominantly characterized by dysfunction in the mesolimbic dopamine (DA) system, which plays a primary role in motivated behaviors [32]. Five dopa-mine receptors have been identified and classified into D1-like families containing D1 and D5 receptors, and D2-like families including D2, D3, and D4 receptors [34]. The D1-like family induces an increase in cyclic AMP synthesis (cAMP) and dopaminergic neuron activity, while the D2-like family inhibits cAMP synthesis, which correlates with the decreased activity of dopaminergic neurons [35]. Animal studies indicate that anhedonia is functionally associated with a decreased activity of mesolimbic dopamine projections from the ventral teg-mental area (VTA) and glutamatergic transmission from the ventromedial prefrontal cortex (PFC) to the nucleus accumbens (NAc) in the ventral striatum [36]. There are multiple reciprocal interconnections between different subregions of the PFC, VTA, NAc, amygdala, and hippocampus, suggesting that these circuits mediate in the behavioral response and development of depression [32]. The hippocampus and PFC play important roles in regulating mood, cognitive function, anxiety, and fear. These structures are the most sensitive brain region to the neurotoxic effects of GCs [33, 37]. The clinical studies in both healthy and depressed humans show that hippo-campal-dependent learning appears to be more sensitive to the effects of cortisol [38]. Additionally, Rocchetti et al. [34] have shown that genetic deletion or the pharmacological blockade of presynaptic D2 receptors in dopaminergic fibres of the temporal hippocampus led to the inhibition of long-term depression (LTD) and play a major role in regulating hippocampal learning and memory. Chronic GC administration also decreased volume, spine density, and reduced activity in the prefrontal cortex, similar to changes observed in depressed patients [39, 40].
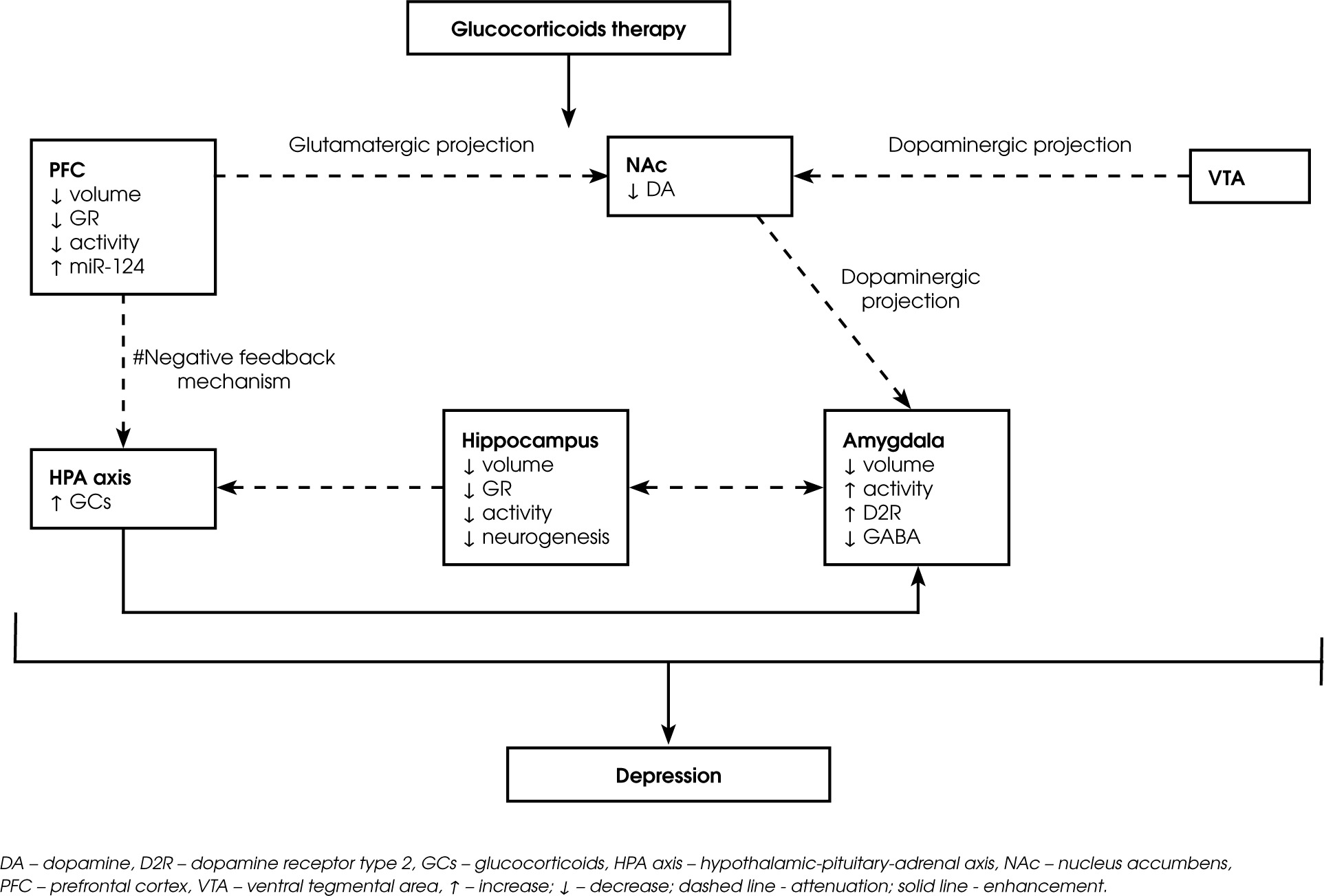
The amygdala is a brain structure involved in the modulation of emotional processes and HPA axis activity, and GCs play an important role in regulating these processes [41-43]. In humans, higher cortisol levels while viewing emotionally arousing pictures were associated with greater amygdala activation in functional magnetic resonance imaging (MRI) [41]. Furthermore, patients receiving chronic GCs therapy had smaller amygdala volumes than control subjects [43]. The postsynaptic D2 receptors primarily residing on GABAergic neurons modulate the activity of the amygdala [44]. An increase in dopamine D2 receptor activity in the amygdala attenuates the local inhibitory function, leading to the enhanced neural activity associated with a negative emotional state [45, 46]. Furthermore, corticosterone treatment enhances the excitability of principal basolateral amygdala (BLA) cells by increasing their intrinsic excitability and decreasing the impact of GABA-A receptor inhibition [42]. Thus, it seems that corticosterone changes in local dopamine D2 receptors activity coregulate the activity of the GABAergic system in the amygdala. Figure I shows the role of microRNAs and the dopaminergic system in the possible mechanism of GC-induced depression.
Figure I
Schematic illustrates the role of microRNAs and the dopaminergic system in the development of depression in subjects subjected to GCs therapy. Long-term GCs therapy leads to a decrease in the activity of the PFC and hippocampus, which consequently impairs the feedback mechanism of the HPA axis and the sustained increase in cortisol. Chronic GCs therapy also attenuates dopaminergic projection from the VTA and glutamatergic projection from the PFC to the NAc, leading to diminished DA release. In addition, increased activity of D2Rs in the amygdala, which lowers the function of the GABAergic system, leading to higher local neuronal activity and enhanced negative emotional state. GCs therapy also induces changes in microRNA expression (e.g. miR-124), which exacerbates depressive symptoms
PHARMACOLOGICAL TREATMENT OF GLUCOCORTICOIDS-INDUCED DEPRESSION
The available data concerning the effects of antidepressants in patients taking corticosteroids is limited and mixed. Initial treatment of patients with GC-induced depression with tricyclic antidepressants (TCAs) was associated with increased agitation and psychosis [47]. However, studies also show TCA’s efficacy in GC-induced depression [48]. The use of selective serotonin inhibitors (sertraline, fluoxetine, or fluvoxamine) was effective in treating depressive symptoms associated with GCs use [49-51]. Additionally, administration of olanzapine (an atypical antipsychotic, multi-target block drug) during GC therapy was successful in the treatment of subacute mood changes [5, 52].
CONClUSIONS
Synthetic GCs, due to the broad spectrum of their activity, are used in many diseases, but they often cause adverse effects, including depression. The mechanism of GC-induced depression is unclear. Available data suggest that the dopaminergic system impaired by long-term GC administration and epigenetic mechanisms, especially microRNAs, may play an important role in these processes. Special attention is directed to the search for specific changes in microRNA expression that will provide clinically practical and easily applicable biomarkers of depression risk and new forms of pharmacotherapy in GC-induced depression.






