






Introduction
Cutaneous lupus erythematosus (CLE) and polymorphic light eruption (PLE) are photosensitive cutaneous manifestations in both diseases. It can be difficult to differentiate histopathologic grounds especially at the onset of both diseases. It cannot help in direct and indirect immunofluorescence [1]. ANA may be useful to distinguish CLE from PLE patients, but ANA is present in some PLE patients [2]. Dermal mucin deposition and marked dermal oedema were found to be common histopathological findings of both diseases [3, 4]. Some authors have suggested that PLE and CLE share a common pathogenesis. The co-existence of PLE-CLE or possibility of transformation from PLE to CLE is controversial [5–9].
Atypical manifestations of pityriasis rosea (PR) are common in dermatological practice, and the atypicality may be in distribution, course, morphology, size, or symptoms [10]. Therefore, it may even cause a diagnostic challenge. Pityriasis rosea patients whose symptoms last more than six months are considered as persistence. Prolonged PR patients with the atypical course should be distinguished from CLE [11].
Mycosis fungoides (MF), which is the most common cutaneous T cell lymphoma type, is an indolent and slowly progressive disease. Its incidence rate in the US is approximately 0.4 cases per 105 with the dominance of the male gender [12]. It is commonly distributed over sun-protective areas depending on the antigenic stimulation of T cells. Discoid, atopic, contact and chronic superficial scaly dermatitis should be considered in the clinical differential diagnosis. On the other hand, histopathologically, it may cause a diagnostic challenge in early stages due to the showing of spongiotic, lichenoid, or psoriasiform major tissue reaction patterns [13].
Plasmacytoid dendritic cells (PDC) are not found in normal skin [1]. PDCs are a dendritic cell subtype with an important function in the production of type I interferons (α/β). These are important in the initiation of inflammation in the pathogenesis of autoimmune diseases, cutaneous malignancies, skin infections, and immune-allergic dermatoses [14]. Type 1 interferon plays an important role in the pathogenesis of lupus erythematosus and is generally produced by PDCs. PDCs can be active by interleukin-3 [15].
PDCs can be marked with CD123. The importance of CD123+ PDC in CLE, cutaneous neoplasms and other inflammatory dermatoses has been investigated in recent years. Studies have involved rosacea [16], lichen planopilaris [13], central centrifugal cicatricial alopecia [17, 18], mycosis fungoides, subcutaneous panniculitis-like T cell lymphoma [19, 20], squamous cell carcinoma and actinic keratosis [21].
Aim
This study investigated the presence and distribution pattern of CD123 + PDC in patients with CLE, MF, PR, and PLE. Our aim was to determine the importance of CD123 + PDCs in differentiating CLE patients from MF, PR, and PLE patients whom we could not distinguish histopathologically.
Material and methods
Appropriate permission for the study was obtained from the Ethics Committee of Duzce University Medical Faculty (approval no. 2018/06). The pathology database of Duzce University Department of Pathology was searched for 20 years, and all cases were identified. Haematoxylin and eosin-stained slides were reviewed by two of the authors (MG, SB) to confirm the diagnosis. The inclusion criteria were 1) diagnosis of MF, PR, PLE or CLE, 2) sufficient clinical history and findings, and 3) sufficient pathology material for analysis. We excluded 21 patients without sufficient paraffin tissue or clinical information from the initial patient group. Therefore, a total of 76 cases (MF (n = 27), PR (n = 19), PLE (n = 11), and CLE (n = 19)) had sufficient clinical information and pathology material and constituted the study group. The clinical features (gender, mean age, biopsy localization) were obtained from the database. The histopathological features were also recorded as follows: acanthosis, spongiosis, parakeratosis, basal cell vacuolization, apoptotic keratinocytes, density of lymphocytic infiltration, epidermotropism, Pautrier microabscess, folliculotropism, angiocentricity, syringotropism, cerebriform nucleus, perinuclear halo, nuclear hyperchromasia, dermal fibroplasia, pigment incontinence and lymphocyte size in MF cases; vacuolar change, dyskeratosis, increased dermal mucin, pigment incontinence, follicular plug, thickened basal membrane, superficial-deep and perivascular-periadnexal lymphocytic infiltration and plasma cells in CLE cases; acanthosis, spongiosis, parakeratosis, erythrocyte extravasation, papillary dermal oedema and superficial lymphocytic infiltration in PR and acanthosis, spongiosis, parakeratosis, erythrocyte extravasation, papillary dermal oedema and superficial and deep lymphocytic infiltration in PLE cases.
Five micron thick sections from each case’s formalin-fixed paraffin-embedded samples were immunostained using the primary antibody against CD123 (dilution 1 : 100, clone BR4MS, UK) with an avidin-biotin-peroxidase technique for the presence of PDC. Clusters of CD123+ PDCs were defined as tight aggregates of ≥ 15 CD123+ PDCs. The percentage of immunolabelled cells of inflammatory infiltrate was counted semi-quantitatively, and the pattern of the staining was noted as previously described.
Results
The mean age at diagnosis of MF, CLE, PR, and PLE was 55.4; 53.9; 31.6 and 36.3 years, respectively. While female gender was dominant in MF (M/F = 13/14) and PR (M/F = 7/12), male dominance was seen in CLE (M/F = 10/9) and PLE (M/F = 6/5). The most common biopsy sites were the trunk for MF (44.4%) and PR (47.3%), head and neck for CLE (63.1%) and PLE (45.4%).
Erythrocyte extravasation (100%), acanthosis (100%), spongiosis (100%), parakeratosis (100%) were seen in PR cases, whereas papillary dermal oedema (100%) was predominantly present in PLE cases. Superficial and deep lymphocytic infiltration was seen in PLE cases; besides, there was no dermal mucin. The lymphocytes of MF cases showed nuclear enlargement, hyperchromasia, cerebriform features, epidermotropism (mostly in the lower to the middle epidermis). Besides, folliculotropism (18.5%), angiocentricity (22.2%), syringotropism (11.1%) and dermal fibroplasia (81.4%) were present in some MF cases. We observed basal cell vacuolization (100%), pigment incontinence (84.2%), and apoptotic keratinocytes (47.3%) in CLE cases. Thickened basal membrane was seen in all CLE cases; also increased dermal mucin (63.1%) and follicular plug (42.1%) were common histopathological findings. Available immunofluorescence results of cases also showed concordance with the diagnostic entities (negative for PLE and PR; a granular deposition of Ig and C3 complement at the dermo-epidermal junction for CLE; not available for MF cases). In all cases, aberrant CD123 immunoexpression has been observed in keratinocytes. Clinicopathological features and CD123 status of the included entities are summarized in Table 1. Histopathological features and immunostaining of CD123 are shown in Figures 1 A–H.
Table 1
Clinicopathological features and CD123 immunoexpression of all cases
Figure 1
A – Minimal spongiosis, erythrocyte extravasation and perivascular lymphocytic infiltration in a PR case (H + E, 200×). B – CD123 immunoexpression in a PR case (200×). C – Superficial and deep perivascular/periadnexal lymphocytic infiltration in a DLE case (H + E, 40×). D – CD123 positive plasmacytoid dendritic cell clusters in a DLE case (200×). E – Papillary dermal oedema, mild spongiosis and perivascular lymphocytic infiltration in a PLE case (H + E, 200×). F – CD123 positive plasmacytoid dendritic cell clusters in a PLE case (200×). G – Atypical lymphocytic infiltration showing epidermotropism in a MF case (H + E, 200×). H – Atypical lymphocytes showed negative for CD123 in a MF case (200×)
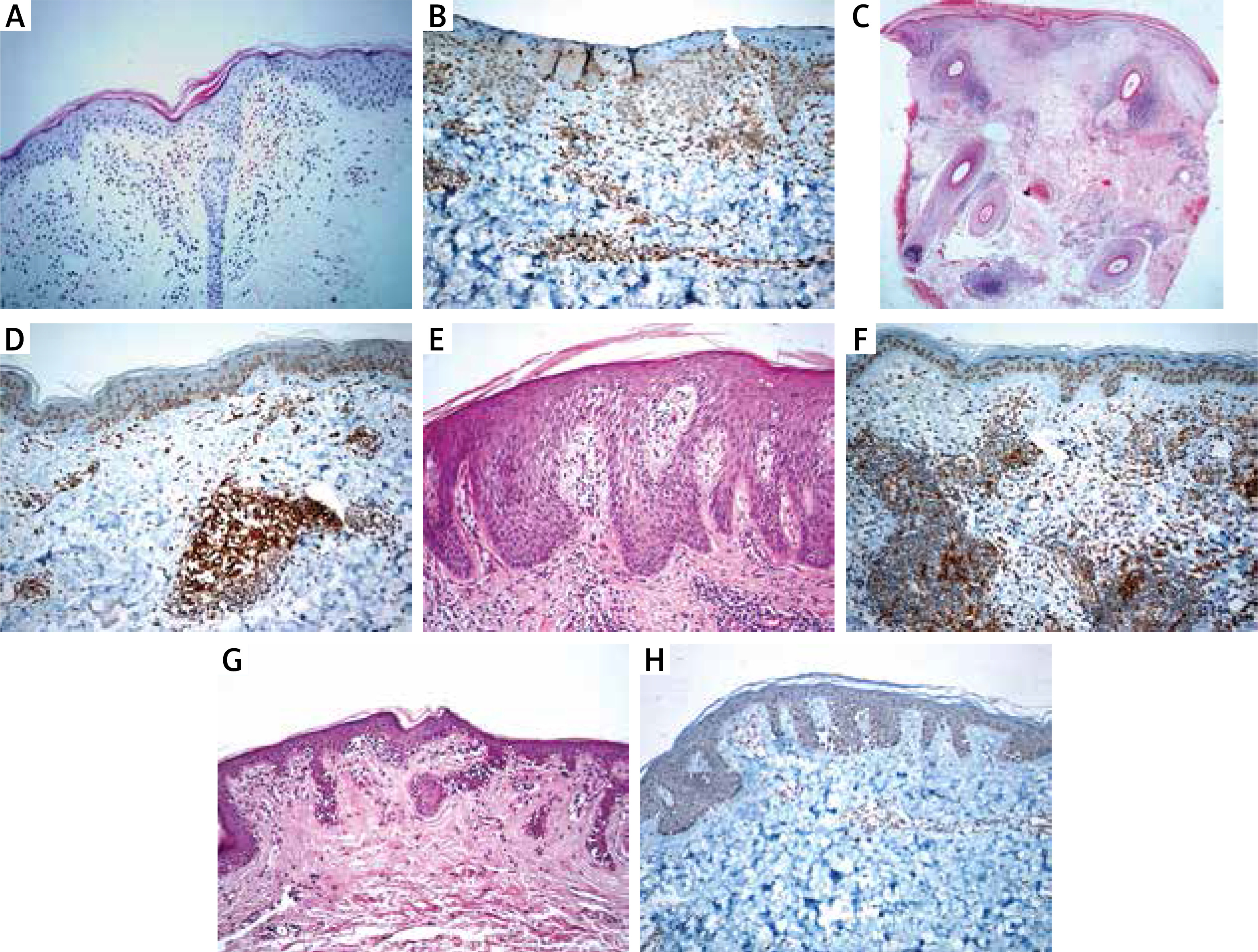
PDC immunolabelled with CD123 antibody was present in MF, PR, PLE, and CLE cases with a mean percentage of 2%, 8.8%, 15%, and 15.2%, respectively. The clustering of PDC was a distinctive feature of CLE when compared with MF (p < 0.001) and PR (p < 0.001); however, PLE showed clustering in 45.5% of cases, which was not significant in the differential diagnosis of CLE (p = 0.612).
Discussion
PDC is a subset of dendritic cells that produce type I interferon (α/β). PDC has CD123 (interleukin-3 receptor chain) or BDCA-2/CD303 surface antigens demonstrated by immunohistochemistry. Type 1 interferon produced by PDCs has a critical role in the onset, progression, and activation of autoimmune and immuno-allergic dermatoses, cutaneous neoplasms, and infectious diseases [13, 19, 22, 23]. Surface expression of interleukin-3 receptor α chain (CD123), which is easily demonstrated by immunohistochemical stains, is used in the differential diagnosis of LE cases to distinguish them from inflammatory/autoimmune diseases [24–27]. The number and distribution of PDCs also differ markedly in inflammatory and neoplastic diseases. Clustered PDCs are a more expected finding than single cells in cutaneous lupus patients [15]. Lichen planus, alopecia areata and psoriasis early plaque lesions have been detected. PDC infiltration in vitiligo patients may be an early indicator of disease progression due to IFN-γ production, and contact eczema has been detected anecdotally [25]. Besides, PDC infiltration was detected in pityriasis lichenoides [28], squamous cell carcinoma-keratoacanthoma [29], molluscum contagiosum [30], and Kaposi sarcoma [31]. Low PDC levels in patients with lupus erythematosus panniculitis (LEP) have been reported to reduce the response to treatment and be valuable in the prognosis of the disease [32].
Although the most cases of CLE are readily distinguishable from MF, PR and PLE, however, sometimes the differential diagnosis can be challenging. To distinguish CLE and MF may be difficult in H + E stained slides, with the presence of interphase dermatitis or necrotic dyskeratotic keratinocytes in MF [19, 33–35] or atypical epidermotropic lymphocyte infiltration with cerebriform nuclei and band-like lymphocytic infiltrate in dermo-epidermal junction in CLE [19, 36].
Previous two studies of Pileri et al. showed that PDC was rare in MF patients, but when the stages were compared, it is significantly higher in stage IIB than stage IA/B disease [37]. The cluster of CD123+ PDC was detected in 3 of 6 patients with granulomatous MF and it implies CD123 immunoreactivity could be identified in granulomatous MF [24]. Additionally, recent studies showed that CD123+ PDCs tend to contribute to cutaneous granulomatous disorders such as granuloma annulare, granulomatous foreign body reactions, and even cutaneous sarcoidosis [38–40].
In a study comparing CD123+ cell infiltration with Jessner’s lymphocytic infiltrate and CLE, there was no statistically significant difference [41]. Chen et al. found CD123+ cell infiltration helpful to differentiate CLE from MF and LEP from subcutaneous panniculitis-like T-cell lymphoma (SPTCL) [19]. Liau et al. also reported that CD123+ cell infiltration might be useful to distinguish LEP from SPTCL [20]. We have similar findings with other studies in the literature, and CD123 immunoreactivity in PDCs seems to be a useful immunohistochemical marker in the differential diagnosis of CLE and MF.
It may be difficult to differentiate LE and PLE histopathologically, and this makes it more difficult to distinguish the two diseases at onset [14]. There might be a stronger relationship between PLE and CLE than previously reported, and PLE susceptibility may contribute to the development of CLE [42]. Nyberg et al. increased the/noted an increased prevalence of PLE in LE patients, a history of PLE in 50% of their patients with CLE, and 33% of those with subacute cutaneous LE (SCLE) [5]. Millard et al. found that 49 of 85 SCLE-CLE patients had PLE history, and 55 of 135 PLE patients had LE in their close relatives [6]. Additionally, 208 PLE patients did not show LE transition in long-term follow-up [7, 8]. Molina-Ruiz et al. [43], Wackernagel et al. [44] and Lei et al. [45] found no CD123+ PDCs in skin biopsies of PLE patients. In contrast to these studies, Rossi et al. claimed PDCS may play a significant role in the development of PLE, and PLE skin biopsies showed that the dermal distribution of PDCs promotes possible association with CLE [46]. PLE and LE are photosensitive diseases, and UV-B exposure exacerbates the clinical findings of both diseases [1]. The results of two previous studies investigating the effect of UV-B exposure on CD123+ PDCs infiltration on the skin surface in mice were controversial; Yin et al. reported CD123+ PDCs accumulation [47], and Sontheimer et al. [48] did not. In our study, there was no statistically significant difference in PDC infiltration and distribution in PLE and CLE patients, and the findings supported Rossi et al.’s study. Similar histopathological features as mentioned above may support the relationship between PLE and LE.
PR atypical eruption may vary morphologically (vesicular, purpuric, haemorrhagic, urticarial, erythema multiform-like lesions) and in contrast to classical PR, the facial, axilla, groin areas, penis and oral cavity, are more frequently involved [11, 49, 50]. Histopathological findings in PR are not pathognomonic for the disorder. Persistent PR is an atypical form that is present for more than 6 months [50]. Rash in PR patients at an unusual location must be distinguished from other dermatoses if the duration of the disease is long. In our study, we assessed the patients with PR who have an atypical rash. Cluster CD123+ PDCs in PR patients were detected in 1/19 patients. Cluster CD123+ PDCs are a prominent histopathologic finding in LE patients [15, 19]. Cluster CD123+ PDCs were found to be 14/19 positive in CLE patients. These results show that cluster CD123+ PDCs can be helpful in the differential diagnosis of CLE patients from PR patients.
The most important limitation of the study was the comparison of the small number of patient groups. Clinical follow-up data and files of patients diagnosed with CLE could not be obtained sufficiently. Another limitation is that TCR gene rearrangement was only studied in a small subset of MF cases at the time of diagnosis.
Conclusions
Our findings suggest that CD123 immunohistochemical staining may be useful to differentiate CLE from MF and PR. There was no statistically significant difference in PDC infiltration and distribution between PLE and CLE patients. Especially, CD123+ PDC clusters seem to be highly specific for CLE and PLE. We suggest that we support the relationship between PLE-LE by demonstrating histopathological similarities. Although immunohistochemical staining with CD123 showed high specificity and sensitivity in comparison with histopathological features in CLE and PLE, we think that clinical history, histopathological evaluation, other immunohistochemical and laboratory findings should be interpreted together to diagnose CLE and PLE.