
Dystrophic epidermolysis bullosa (DEB) is a genetic mechanobullous skin disorder that manifests at birth or in early infancy. The hallmarks of DEB are blister formation, skin fragility, and nail dystrophy following minor trauma. The disorder results from mutations in the type VII collagen gene (COL7A1) encoding the type VII collagen protein (C7). C7 is a major component of anchoring fibrils (AFs) [1], which is critical for attachment of the epidermis to the dermis. Dysfunction or loss-of-function of C7 leads to DEB. For instance, the complete loss of C7 causes the Hallopeau-Siemens type of DEB – the most severe phenotype. The inheritance pattern of mutated COL7A1 is either autosomal dominant (DDEB, OMIM 131750) or autosomal recessive (RDEB, OMIM 226600). However, de novo spontaneous mutations of COL7A1 are rarely reported in the population. Herein, we describe a DEB patient with a mild phenotype caused by a de novo missense mutation in the amino-terminal non-collagenous (NC)1 domain of C7.
The proband was a 4-year-old female single child born to non-consanguineous parents without any family history of blistering disease. The patient visited the clinic of the Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College, Nanjing, China in September 2017. The chief complaint was blisters in response to skin trauma and dystrophic toenails since birth. Her parents were free of symptoms of DEB, but the child suffered from blistering limited to trauma-exposed sites and toenail dystrophy. At the time of the medical examination, the skin sites were healed and covered with atrophic scars. No other malformation of the patient was detected.
In order to determine the pathogenesis, we collected the peripheral blood from the patient, and extracted DNA from lymphocytes for sequencing. We sequenced all 118 exons, exon-intron borders, and the promoter region of COL7A1 using PCR amplicons according to a published paper [2]. Sequencing results and mutational analysis revealed a heterozygous mutation of C to T in the COL7A1 gene, located at position 2183 of exon 17 (Figure 1). No other known mutations in the COL7A1 gene were found. Further analyses revealed that the mutation resulted in the S728F variation in protein C7. The mutation was not detected in the patient’s parents, indicating the mutation was indeed de novo. More importantly, we sequenced 100 random healthy volunteers and failed to find this mutation. By searching a genome database, gnomAD (http://gnomad.broadinstitute.org/) [3], we were able to confirm that this mutation had not been reported.
Figure 1
Sequencing results of the mutation and predicted variation in C7. A – Heterozygous mutation of 2183C>T of COL7A1 (the top panel). The sequence of the same region in a health volunteer (the lower panel). B – Overall view of the predicted structure of the NC1 domain of wild-type C7, with the β-sheets in red and the loops in pink. C – Local view showing the hydrogen bond (green dotted line) between Ser718 and Ser728 of wild-type C7. D – Partial view showing the mutant domain with Phe728 and the loss of the hydrogen bond
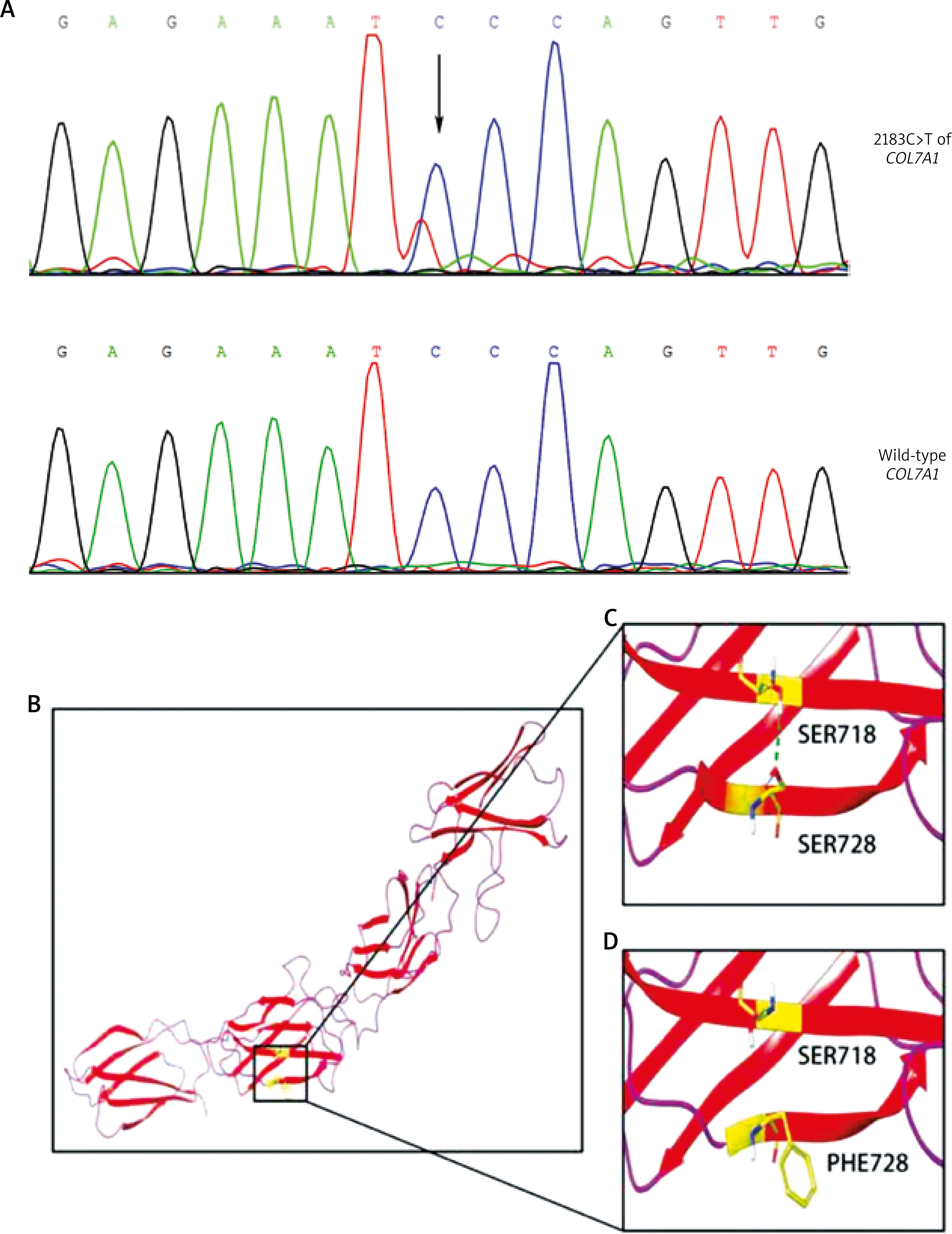
By taking the advantages of existing databases, we sought the biological significance of the mutation. The 2183C>T mutation has been predicted to be detrimental by PolyPhen-2 (score = 0.05) (http://genetics.bwh.harvard.edu/pph2/) [4] and CADD (Phred-score = 23.8) (http://cadd.gs.washington.edu/snv). Furthermore, we used SWISS-MODEL website [5] and Maestro software [6], respectively, to predict the 3D model of C7 (SMTL id: 1fnf.1) and observe the structure related to amino acid residue 728. We found that the hydrogen bond between the side chain of Ser728 and Ser718 in the wild type collagen was broken in the mutated protein, probably causing protein instability (Figure 1).
In addition to advice on standard skin care for the patient, we also suggested that the parents take genetic consultation during the patient’s child bearing ages. The patient has been followed up since discharge (September 2017), during which the symptoms have not exacerbated.
DEB is a group of genetic conditions whose hallmarks are skin fragility and nail deformity. The pathogenesis of DEB is mutations in COL7A1 leading to dysfunctional C7 or a completely loss of C7. C7 acts as a key member of AF, residing underneath the lamina densa in the papillary dermis. C7 connects to laminin-332 and type IV collagen in the epidermal basement membrane through C7’s NC1 domain [7], and interacts with type I collagen in the dermis [8]. C7 is a homotrimer that is comprised of three identical α unit chains. Each α chain is composed of a 145-kDa central collagenous triple-helical domain (TH) with the characteristic repeating Gly-X-Y amino acid sequence. A 145-kDa amino terminal, named NC1, and a 34-kDa carboxyl-terminal NC2 flanks the TH domain.
Although it has been reported that approximately 600 mutations in the COL7A1 gene cause DEB, there are fifteen missense mutations found within the NC1 domain (Table 1) [9]. There is only one missense mutation reported within the NC1 domain causing DDEB. This mutation is widely considered to be associated with glycine substitutions in the TH domain of C7. As the second identified missense mutation to cause DDEB within the NC1 domain, S728F mutation is also the first in the fourth-last fibronectin type III-like repeat, which results in a mild disease phenotype.
Table 1
Phenotypes and genotypes of DEB patients with missense mutation in NC1 domain of the COL7A1 gene [9]
We were unable to determine the biological effects of the S728F mutation, which require multidisciplinary collaborations. Given a predicted loss of the hydrogen bond between Ser728 and Ser718, it is reasonable to speculate that the mutation causes impaired stability of C7. Also, the probability cannot be excluded that the S728F mutation might interfere in the structure of the TH domain or the interaction between the NC1 domain and extracellular matrix components.
In conclusion, the de novo missense mutation S728F observed in the NC1 domain of C7 from this patient may result in a relatively mild phenotype. However, the prevalence and genotype-phenotype correlation of this mutation in the NC1 domain should be explored further.







