
INTRODUCTION
The most common genetically determined small vessel disease associated with strokes and vascular dementia is CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, OMIM #125310) [1, 2]. CADASIL is caused by a mutation in NOTCH3 gene, which consists of 33 exons located on the short arm of chromosome 19 (19p13.1) [3] and encodes a single-pass transmembrane receptor predo-minantly expressed in vascular smooth muscle cells in adults. Typical mutations include cysteine-related missense mutations located in one of the 34 EGF-like repeats (EGFr) in the NOTCH3 receptor [3]. Almost 600-point mutations in this gene have been identified so far (The Human Gene Mutation Database [HGMD] database, as of 02/04/2025) [4].
The core magnetic resonance imaging (MRI) abnormalities observed in CADASIL include white matter hyperintensities, subcortical infarcts and cerebral micro-bleeds [5]. The radiological presentation of CADASIL varies considerably between the affected individuals [6]. MRI findings were observed in most patients aged > 35 years [7]. Migraine with aura (MA) in younger patients, acute encephalopathy, recurrent lacunar strokes, cognitive decline, mood and gait disturbances in older patients are typical clinical manifestations of CADASIL [8]. Diagnosis mostly coincides with the onset of stroke without vascular risk factors and subcortical dementia between the fourth and sixth decades [8].
Beginning with the first description of CADASIL, MA has been detected extremely frequently among pre- symptomatic family members and is now considered a classic phenotypic feature of the disease [7, 8]. In contrast to MA, migraine without aura (MO) was not found to be more prevalent in CADASIL than in other large population-based samples [9, 10]. Therefore, migraine absence or only MO in CADASIL patients, especially within the same family, may result in a missed diagnosis. To highlight the problem of phenotypic variability, we present the case of a three-person family with a pathogenic NOTCH3 variant.
Case description
A 67-year-old female patient (II 2; Figure IA) was hospitalized for a sudden onset of right hemiparesis (Medical Research Council [MRS] 4/5) and speech impairment. She was not eligible for thrombolysis owing to the arrival at the hospital after the recommended therapeutic window. The patient’s neurological condition remained stable during hospitalization.
Figure I
A) A family tree of a family with a NOTCH3 gene mutation; B, C, D) FLAIR MRI sequences of individual family members: B – II2, C – II3, D – III-1; the arrows indicate small-vessel vascular changes. A detailed description is provided in the text
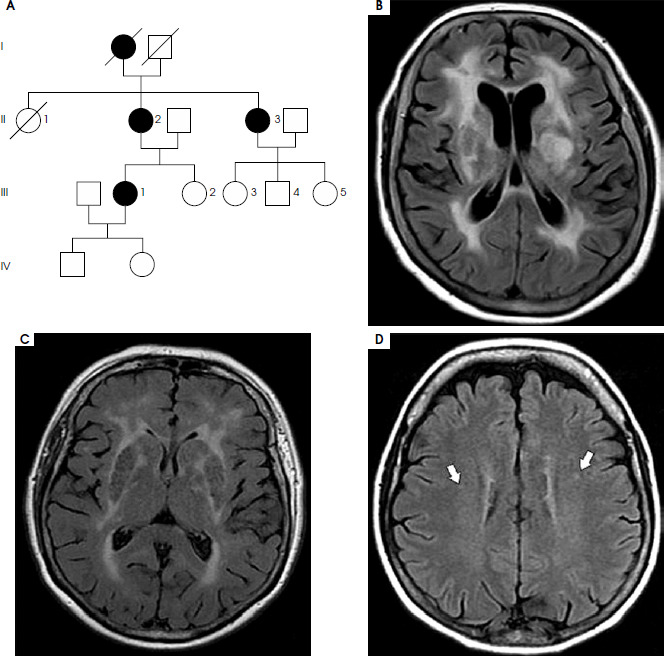
There were no hemodynamically significant abnormalities in carotid flow, atrial fibrillation, or changes in the cardiac cavities, which could be a potential cause of ischemic stroke. Baseline and follow-up brain computed tomography did not show any abnormalities, but MRI revealed diffuse white matter changes with involvement of the temporal lobes (Figure IB) and microbleeds in subcortical nuclei. Considering the radiological findings and family history of dementia, we suspected CADASIL. Genetic testing identified a rare heterozygous variant of NOTCH3 gene NM_000435:c1675T>G p.(Cys559Gly). The variant was classified as likely pathogenic according to the American College of Medical Genetics and Geno-mics (ACMG)-AMP criteria [11], indicating > 90% probability of pathogenicity (applied criteria: PM1, PM2, PP3_Strong). It involves a transversion of a highly conserved nucleotide (PhyloP100 score = 9.25) in exon 11 of 33 of the NOTCH3 gene, resulting in a missense substitution of cysteine to glycine at codon 559 of the polypeptide chain. The NOTCH3 gene is sensitive to missense variation (gnomAD v2.1.1 Z-score = 3.53), which denotes gene-level depletion (fewer than statistically expected) of missense variants. The region encompassing the variant (residues Gly140-Glu535) exhibits significant genomic constraint against missense changes (p = 1.694 × 10–4), indicating a subgenic segment with significant intolerance to missense changes; missense variants arising within it are therefore more likely to be deleterious than the variants elsewhere in the gene. Both metrics support the variants’ pathogenicity in this genomic interval. The variant was absent from control chromosomes in gnomAD (v4.1.0).
To confirm pathogenicity, other family members were evaluated, both clinically and genetically. The proband’s 65-year-old sister (II 3; Figure IA) was diagnosed with dementia (Mini-Mental State Examination [MMSE] score: 15 points). MRI showed diffuse white matter involvement with temporal lobe damage (Figure IC) without subcortical microbleeds. Genetic testing confirmed the presence of the same likely pathogenic variant: c.1675T>G p. (Cys559Gly).
The 43-year-old daughter of the proband (III 1; Figure IA) had experienced headaches meeting the diagnostic criteria for MO according to ICHD-3 since high school. Her neurological examination was normal. Only the brain MRI revealed a disseminated small vessel lesion (Figure ID). Genetic testing confirmed the occurrence of the same variant in her mother and in her mother’s sister.
Discussion
A new, previously undescribed likely pathogenic he-terozygous variant was found c.1675 T>G p.(Cys559Gly) (pathogenicity assessment according to ACMG – likely pathogenic variant – class IV) consistent with the CADASIL phenotype. Its pathogenicity is supported by a correlation with the typical CADASIL phenotype, which features in the proband (II 2) and her sister (II 3). In both cases (II 2, II 3), dementia was diagnosed and diffuse white matter changes were found on MRI. In addition, the proband (II2) developed cerebral microbleeds – a common finding in CADASIL [12].
Another argument for pathogenicity of the reported variant is a previously described likely pathogenic variant in the Chinese CADASIL population, located at the same codon (559) causing a different amino acid substitution: c1677 C>G p. (Cys559Typ) [13].
Genetic screening of the proband’s family identified the same variant c.1675C>T p. (Cys559Gly) in one of her daughters (III 1) who complained of a long-standing history of headaches meeting the International Classification of Headache Disorders 3rd edition (ICHD3) criteria for MO [14]. MRI revealed small diffuse ischemic changes in the brain characteristic of small vessel disease but not meeting full CADASIL radiological criteria. In contrast to MA, which is a hallmark feature and well-known first symptom of CADASIL [13-16], MO is less common in patients with mutations in the NOTCH3 gene. In a large French study focused on migraine in CADASIL (N = 378), 54.5% of the participants presented with a positive history of migraine; among them, 84% had MA (45.8% of total cohort) [17], and 10-15% had exclu-sively MO [17].
We believe that the symptoms of small vessel disease observed in the patient (III1), with a confirmed variant c.1675C>T p. (Cys559Gly) represent early radiological signs of the disease. However, due to the less pathogenic type of mutation, characterized by a slower manifestation of vascular changes in the brain, the diagnosis of CADASIL requires pedigree analysis. This family case with a novel NOTCH3 pathologic variant highlights the population of patients with MO and MA with cerebral small vessel disease. Such clinical and radiological overlap has already been described [18-20]. It is likely that in the case of some patients with MO, MA and diffuse vascular changes, we are dealing with early symptoms of CADASIL (or another small vessel disease of different genetic etiology). Knowledge of early diagnostic markers characterizing the prodromal stage of CADASIL may be of great importance when it comes to the early identification of patients eligible for unknown causal treatment.






