
Introduction
Atopic eczema (AE) is the most common chronic inflammatory skin disease frequently associated with other diseases of the atopic diathesis [1]. AE is often considered the window to development of other atopic manifestations, including asthma and allergic rhinitis [2–4]. A multifactorial background for AE has been suggested with genetic as well as environmental factors influencing disease development [5, 6]. Several meta-analyses have confirmed a strong association of the filaggrin (FLG) null mutations with AE and other allergic diseases, especially asthma occurring in association with eczema [7–13]. These results have highlighted that genetically determined impairment of the skin barrier is critical not only for the development of AE, but also for other allergic disorders [14–19]. The evidence from both expression studies and Genome-Wide Association Studies (GWASs) suggest that numerous other genes essential for maintaining the skin barrier are located within the epidermal differentiation complex (EDC), a region on chromosome 1q21, and may be involved in the pathogenesis of AE and progression to secondary allergies similar to the well-known FLG mutations [20–24].
Small proline-rich proteins (SPRRs) encoded on the 1q21 region function as cross-bridging proteins of the cornified envelope (CE) that provides the main barrier function of the skin [25]. Experimental studies have provided data indicating the altered pattern in SPRR protein expression in AE skin lesions and also in a mouse model of asthma [22, 26]. This finding suggests that the SPRR gene may be associated not only with AE, but also with asthma [27–29]. We focused on polymorphisms in the SPRR2 gene because SPRR2 proteins have been shown to be allergen- and IL-13-induced gene products that may be involved in the pathophysiology of allergic responses in diverse mucosal tissues [26].
Aim
In the current study, we aimed to determine the importance of the rs6693927 polymorphism in the SPRR2B gene in the susceptibility to childhood AE and other atopic phenotypes such as eczema-associated asthma. Moreover, we evaluated whether this possible association is independent of the well-described FLG risk alleles.
Material and methods
Study population
A total of 188 unrelated children (107 males) less than 2 years old at the time of recruitment were enrolled; namely, 103 patients with AE (mean age: 13.2 ±6.7 months) and 85 healthy control subjects (mean age: 15.3 ±5.6 months). The whole study population were followed at yearly intervals until the age of 6 years. All study participants were of Caucasian ethnicity. The study subjects were recruited from patients who visited the Outpatient Clinic for Children at the Wroclaw Medical University Hospital and from the general population, as described below. The healthy control subjects were recruited from the general population through community-based approaches. We distributed flyers at local nurseries, child and family doctors’ surgeries, health fairs. Interested parents were instructed to contact the research coordinator by telephone. All participants, cases and controls, were selected using a detailed questionnaire that included questions regarding the overall health status, symptoms of AE and other allergic diseases, sociodemographic information and family history of allergic diseases. The subjects with AE were examined and diagnosed according to the criteria established by Hanifin and Rajka [30]. The mean age at disease onset was 4.6 ±3.5 months. AE disease severity was assessed by using the SCORing Atopic Dermatitis (SCORAD) index. Subjects with AE were divided into allergic and non-allergic on the basis of the positive specific IgE level against at least one of the allergens tested. Asthma was defined by the presence of one or more wheezing episodes during the previous 12 months at the age 5 and/or 6 years or a physician’s diagnosis of asthma by 6 years of age. Allergic rhinitis was defined by a physician’s diagnosis of allergic rhinitis by 6 years of age. The control group included healthy children and met the following criteria: absence of symptoms of AE and asthma or allergic rhinitis and negative family history of allergic diseases.
Serum measurements for total and specific IgE levels were performed in all recruited subjects, including IgE specific to the most popular 10 inhalant and 10 food allergens. The levels of specific IgE were determined using a standard enzyme immunoassay (Polycheck, BIOCHECK, Germany). Allergic sensitisation was defined as the presence of a specific IgE to at least one of the tested allergens of ≥ 0.7 kU/l (Class II).
The study was approved by the Ethics Committee of the Wroclaw Medical University, Wroclaw, Poland and informed written consent, including consent to genetic studies, was obtained from all of the subjects before testing.
Genotyping
Samples of the 188 subjects were genotyped for the SPRR2B rs6693927 SNP and for the four common FLG mutations: R501X, 2282del4, R2447X and S3247X on chromosome 1q21 as described previously [31, 32]. Genomic DNA was obtained from EDTA whole blood samples using the QIAamp DNA Blood Mini Kit (QIAGEN GmbH, Germany). All mutations were determined using a LightSNiP assay (TIB Molbiol, Berlin, Germany). PCR was performed in a final volume of 10 μl containing 1 μl of DNA at concentration 15–60 ng/μl, 0.5 μl of reagent mix containing specific primers and SimpleProbe® probes at optimised concentration, 0.8 μl of MgCl2 and 1 μl of LightCycler® FastStart DNA Master HybProbe (Roche Applied Science, Mannheim, Germany). Reactions were performed on a LightCycler 1.5 platform (Roche Applied Science, Mannheim, Germany). For quality control of genotyping procedures, positive controls of each genotype, as well as negative controls, were included in each reaction.
Statistical analysis
The Hardy-Weinberg equilibrium was tested using the χ2 goodness-of-fit test to compare observed genotype frequencies with expected frequencies among the controls. Differences in genotype frequencies or demographic characteristics between case and control groups were evaluated using the χ2 test or Fisher exact test, as appropriate. The associations of genotypes or alleles with patient groups versus control subjects were determined by computing the odds ratio (OR), its 95% confidence interval (95% CI) and p-values, using logistic regression analysis for crude ORs and adjusted ORs when adjusting for family history of atopy. Statistical significance was set at p < 0.05. A χ2 test or Fisher exact test was used to determine the combined effect of genotype pairs. Gene–gene interaction was investigated by using logistic regression models for atopic AE with interaction terms (SPSS). To establish whether an interaction between the two risk factors A (rs6693927) and B (FLG mutations) existed, the relative excess risk due to interaction (RERI), the proportion attributable to interaction (AP), the synergy index (S) and the ratio of ORs were calculated, as recommended by Knoll et al. [33, 34]. Statistical analysis was carried out using the program package Statistica Version 9.0 (StatSoft, Inc., Tulsa, OK, USA) and the SPSS Statistics software package Version 11.1 (SPSS Inc., Chicago, IL, USA).
Results
Baseline characteristics of patients with AE and controls are given in Table 1. There were no significant differences between the cases and controls for age and gender. The allele and genotype distributions for the SPRR2B rs6693927 SNP in the cases and controls are shown in Table 2.
Table 1
Characteristics of the study group
Table 2
Genotype and allele frequencies of SPRR2B rs6693927 in the group studied
| Genotype | Control n (%) | Atopic eczema | Allergic atopic eczema | Non-allergic atopic eczema | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n (%) | P-value | aOR* (95% CI) | n (%) | P-value | aOR* (95% CI) | n (%) | P-value | aOR* (95% CI) | ||
| SPRR2B rs6693927 | ||||||||||
| GG | 32 (38%) | 26 (25%) | 0.152 | 1.00 (Reference) | 18 (33%) | 0.608 | 1.00 (Reference) | 8 (17%) | 0.038 | 1.00 (Reference) |
| AG | 45 (53%) | 62 (60%) | 1.69 (0.84–3.40) | 29 (53%) | 1.14 (0.51–2.57) | 33 (69%) | 2.93 (1.11–7.95) | |||
| AA | 8 (9%) | 15 (15%) | 2.31 (0.76–7.11) | 8 (16%) | 1.78 (0.49–6.42) | 7 (15%) | 3.50 (0.82–15.3) | |||
| AG + AA | 53 (62%) | 77 (75%) | 0.067 | 1.78 (0.91–3.50) | 37 (67%) | 0.553 | 1.24 (0.57–2.69) | 40 (83%) | 0.011 | 3.02 (1.17–8.00) |
| A | 61 (36%) | 92 (44%) | 0.085 | 1.44 (0.93–2.23) | 45 (41%) | 0.397 | 1.23 (0.73–2.08) | 47 (49%) | 0.037 | 1.71 (0.99–2.94) |
In the overall study population, the rs6693927 polymorphism of the SPRR2B gene did not significantly increase the risk of AE; however, there was a trend towards an association of the risk allele A and AE. Moreover, after stratification for allergic and non-allergic AE groups, we found a highly significant association of SNP rs6693927 with AE but only in the non-allergic group. In the logistic regression analysis in the non-allergic group, the rs6693927 AA homozygote and AG heterozygote were associated with a significantly increased risk of AE compared to the wild-type homozygote. The genotype model showed an OR of 2.93 for heterozygote individuals and 3.50 for homozygotes. The OR associated with the rs6693927 SNP was estimated at 1.71, using the allele model (Table 2). The non-allergic and allergic AE groups did not differ in terms of frequency of rs6693927 SNP (p = 0.156; χ 2 = 3.710).
There were no significant associations between AE severity and the SPRR2B rs6693927 SNP. The mild and moderate AE groups did not differ in terms of frequency of SPRR2B rs6693927 SNP (p = 0.971; χ 2 = 0.595; data not shown).
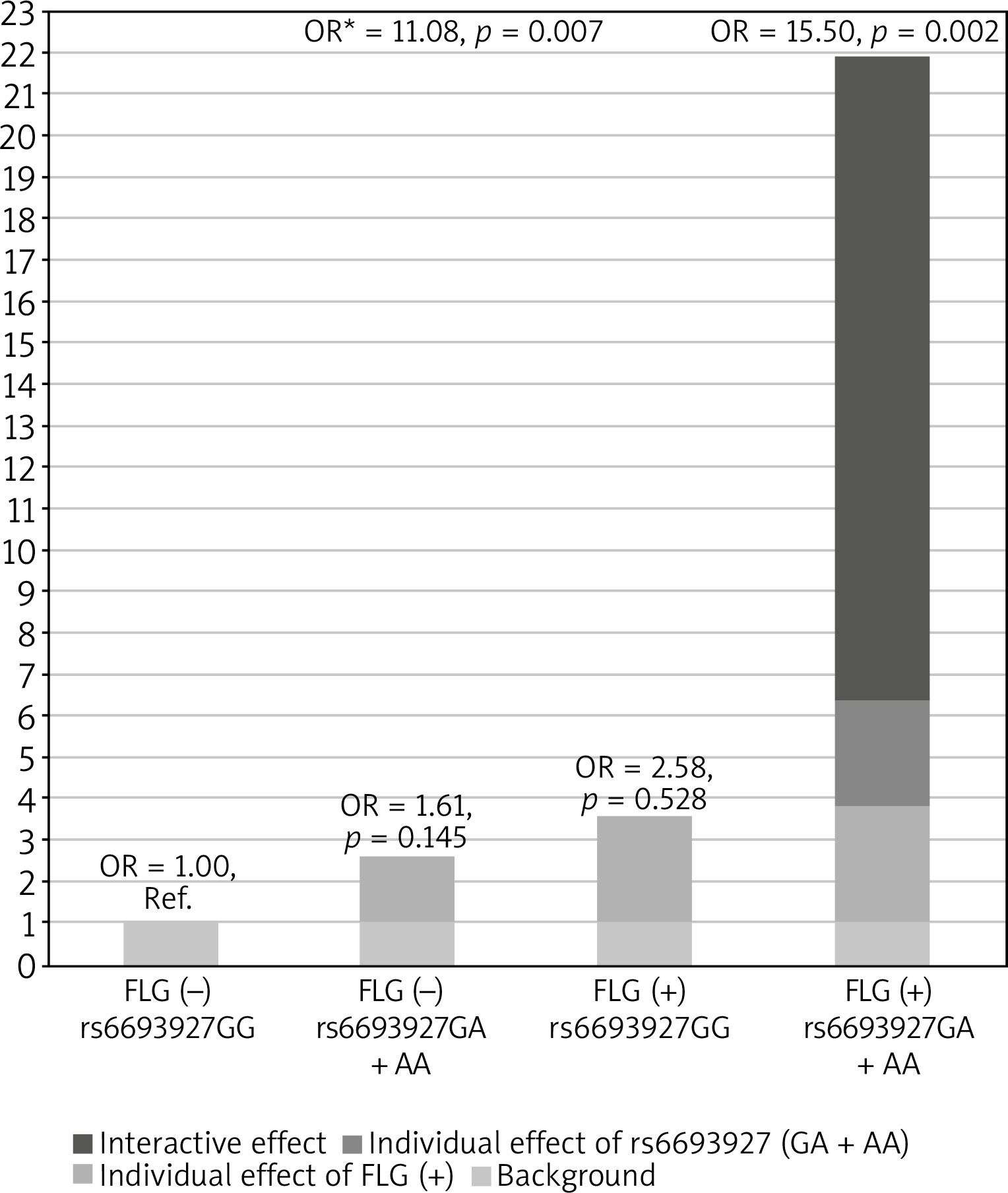
Next, we investigated whether the association of rs6693927 SNP with AE is affected by the strongly significant FLG null mutation status. We therefore included the combined FLG genotype as a second predictor in a logistic regression model to determine whether the FLG mutations confound rs6693927 SNP and AE association in the non-allergic group. After adjusting for the presence of FLG mutations the rs6693927 SNP still showed a statistically significant effect (p = 0.011) with an OR of 3.12 (95% CI: 1.28–7.59). Moreover, we investigated whether an interaction between rs6693927 SNP and the four most common FLG mutations influenced the AE risk. Compared to the reference group carrying neither genetic risk factor, a strong effect was revealed only in subjects who carried both risk factors; the presence of at least one rs6693927 [A] allele and at least one loss-of-function FLG mutation significantly increased the AE risk (Table 3). Figure 1 shows this graphically. Our results suggest that there is some evidence of a gene–gene interaction on additive and multiplicative scales, although when modelling interaction, the interaction coefficient was not significant for both genotype combinations (p-value for interaction = 0.828), which may be because of the low sample size in our study and insufficient statistical power.
Table 3
Interaction between SPRR2B rs6693927 and the FLG mutations in atopic eczema
Finally, we evaluated the association between the tested risk variant and the presence of other atopic phenotypes such as allergic sensitisation, asthma, allergic rhinitis and eczema-associated asthma. In the overall study population, the rs6693927 SNP did not significantly increase the risk of asthma. However, we revealed that the risk allele predisposed to eczema-associated asthma, increasing the risk of this combined phenotype more than 5-fold. In contrast, in the absence of eczema, the association with asthma was not significant. When we then evaluated the rs6693927 SNP as a risk factor for asthma in the subgroup of children with eczema, we could not find an independent effect of rs6693927 on asthma. We did not find an independent effect of rs6693927 SNP on allergic sensitisation and allergic rhinitis in the overall study population and after stratification according to the presence or absence of eczema (Table 4).
Table 4
Associations between SPRR2B rs6693927 genotype and allergic diseases. The control group comprises all individuals, who do not belong to any of the disease group
Discussion
Atopic eczema (AE) is a common inflammatory disease caused by a combination of genetic and environmental factors [5]. The FLG mutations remain the most significant and widely replicated genetic risk factor for AE reported to date [10–12, 35, 36]. These results were also confirmed in our former studies [31, 32] and other study conducted in Polish AE cohorts, which revealed that the FLG mutation, alone and in combination with certain IL-10 or IL-13 polymorphisms, enhances the risk of the development of AE [37]. This is in line with observations that FLG mutations alone are unlikely to account for the total burden of AE at a population level [7, 21, 22, 36, 38]. Recent GWASs for AE have identified an additional association signal in the EDC genes on chromosome 1q21 apart from the mutations in the FLG gene [20, 23, 38–41]. SPRRs, the precursor proteins of CE encoded on the EDC region, are linkers for transglutaminase-mediated protein cross-bridging and play a structural role in all stages of CE formation [25, 42–44]. The experimental studies demonstrated dysregulation in SPRR gene expression accompanied by an altered pattern in the composition of SPRR proteins in patients with AE [22, 45, 46]. In systematic screening for putatively deleterious mutations in the EDC, Marenholz et al. identified an rs28989168 SPRR3 variant that carried an extra 24-bp repeat in the central domain as a previously unreported risk factor for AE [28]. In our study we focused on another SPRR family gene, i.e., the SPRR2B gene, which had previously been found to exhibit changes in expression in inflammatory skin disease and the expression of which is believed to be of major importance for CE biomechanical properties [47–49]. We provided evidence for the highly significant association of rs6693927[A] allele in SPRR2B with AE in the non-allergic group. A weak trend towards an association was also seen in the overall study population. Similarly, Epstein et al. demonstrated a possible association between an rs6693927 SNP in SPRR2B and eczema in the CCAAPS (Cincinnati Childhood Allergy & Air Pollution Study) cohort; however, this finding was not statistically significant [29]. The SPRR2B gene is expressed predominantly in squamous epithelium where it plays an important role in the formation of the insoluble keratinocyte-cornified crosslinked envelope essential for structural integrity and permeability [26, 48]. Accordingly, our results support the hypothesis that SPRR2B variants may influence skin barrier function by altering the physical properties of the CE and thus may contribute to AE susceptibility, confirming the key role of the epidermal barrier in AE development. Interestingly, our study revealed a strong effect of the rs6693927[A] allele on the non-allergic form of AE, whereas there was no effect in the presence of allergic sensitisation. In addition, we demonstrated that allergic sensitisation per se was not associated with the risk allele both in the overall study population and in a subgroup with AE, indicating that IgE-mediated hypersensitivity is not the primary disease manifestation of the risk variant in SPRR2B. It is worth pointing out that the effect of rs6693927 SNP on the AE risk was independent of the well-established FLG null mutations, confirming that other genes located within the EDC, such as SPRR2B, are involved in the pathogenesis of AE and may play an essential role in disease development. Moreover, our results suggest some possible positive interaction on the additive or multiplicative scale between rs6693927 SNP and FLG mutations because the combined disease risk of the both tested variants was significantly higher than that observed for these alleles separately. The interactive effect of SPRR2B and FLG variants is biologically plausible due to the high degree of relationship among the EDC genes, their coordinated expression, as well as the interaction of the encoded proteins in the formation of the epidermis [28, 42].
AE is often the first manifestation of atopic march in early life and often predates the development of allergic airway disease [2]. Based on the assumption that this progression from AE to secondary respiratory allergies is likely to be due to the defective epidermal barrier function, the EDC genes could be predicted to play an important role in asthma development [14–19]. This in particular may apply to the SPRR2B gene due to the potential involvement of SPRR2B proteins in the pathophysiology of allergic responses in diverse mucosal tissues [26]. In our study the compound phenotype of asthma concomitant with eczema has shown a strong, statistically significant association with the rs6693927 risk variant. This is consistent with the study in the CCAAPS birth cohort, which reported that the SPRR2B risk variant is associated with the eczema plus asthma phenotype [29]. Further analysis of the combined allergic phenotypes revealed that the rs6693927 risk variant was associated with asthma only in the context of eczema emphasising an essential role of eczema in the allergic airways disease associated with the rs6693927 risk variant. These results suggest that SPRR2B might predispose to the genetically distinct form of asthma or a particular asthma phenotype occurring in the context of eczema, in contrast to the form of asthma not linked with eczema. However, the mechanism of the asthma risk associated with the SPRR2B variant is not yet fully understood; this may be associated with changes in the biomechanical properties of CE, subsequent skin barrier impairment and percutaneous allergic sensitisation [47–51]. On the other hand SPRR2 expression is not restricted to squamous epithelium [42]. In particular SPRR2B is expressed in lungs of mice, and its expression is specifically altered in the nasal epithelium of asthmatic patients [26, 49, 52]. In situ hybridisation revealed marked allergen-induced expression of SPRR2B in the asthmatic lung, which was observed in a subset of airway epithelial and mononuclear cells associated with allergic inflammation [26]. It has been speculated that allergic lung response that involves epithelial injury and repair may be mediated by the altered expression on the SPRR2 gene as part of a stress response to infection or allergic inflammation signals [26, 49, 53]. These observations raise the possibility that SPRR2B may play an important role in the pathogenesis of the distinct phenotype eczema-associated asthma.
A potential limitation of our study, typical of all case-control studies, is the relatively small sample size and, as a consequence, rather low statistical power, which may lead to false negative or false positive results. Nevertheless, we were able to demonstrate statistically significant and biologically plausible effects. An interaction analysis requires a large population, so our results should be interpreted carefully; there was some evidence of interactions on an additive scale, but there was insufficient statistical power to either confirm or refute these findings.
Conclusions
Our results indicate that the rs6693927 risk variant in the SPRR2B gene may contribute to AE susceptibility, and this effect seems to be independent of the well-established FLG risk alleles, confirming that other genes located within the EDC are involved in the pathogenesis of AE. Moreover, we identified the association of that rs6693927 risk variant with eczema-associated asthma, suggesting that genetically determined impairment of the skin barrier is critical for the development not only of AE, but also for particular asthma phenotype occurring in the context of eczema. In addition, our results revealed that the rs6693927[A] variant and FLG mutations are candidate genes that may control the risk of AE in an interactive manner. We conclude that future studies on AE have to consider gene–gene interaction, as each genetic variant may have their own effect in isolation but the combination of functional SNPs in more than one gene may magnify their impact on the disease.







