
Glucagonoma syndrome consists of the tumour of pancreatic alpha cells and symptoms caused by the abnormal glucagon production. A hallmark sign of this syndrome is necrolytic migratory erythema (NME) first described in 1942 [1]. Its distinctive clinical appearance along with concomitant diabetes mellitus, weight loss, deep vein thrombosis, neuropsychiatric disorders and diarrhoea may suggest the diagnosis. Treatment is curative if the early diagnosis is made and involves tumour surgical resection [2, 3].
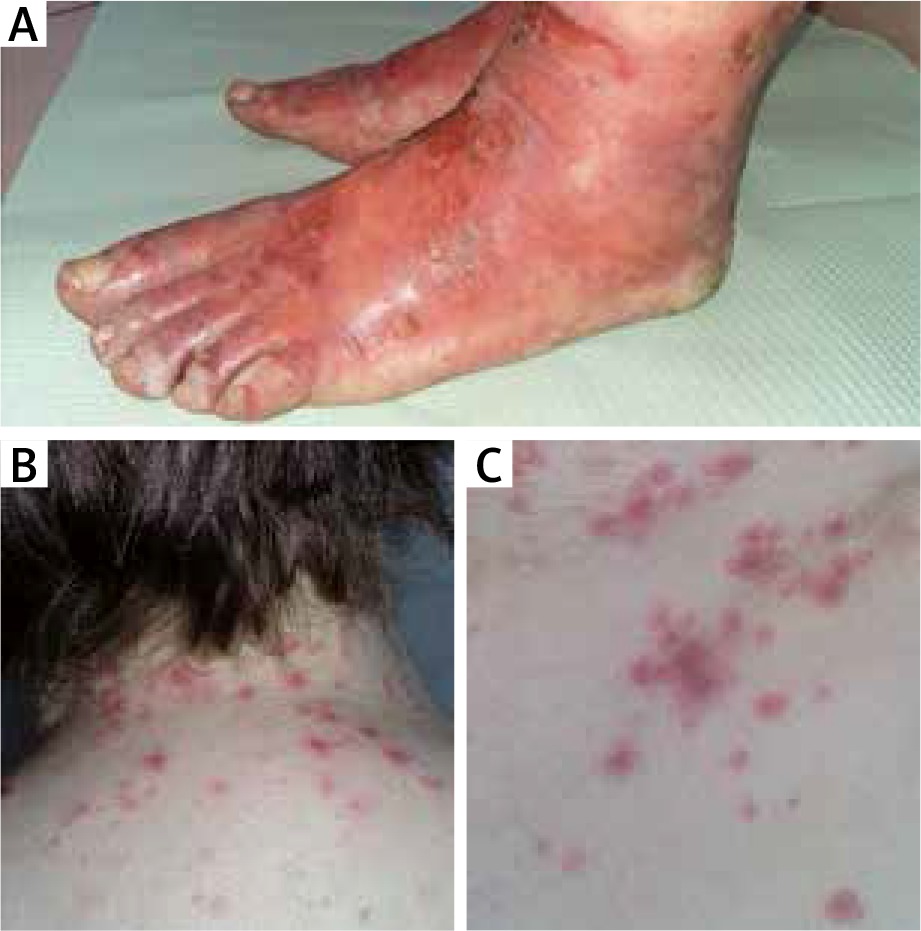
A 49-year-old woman was admitted to the Dermatology Department with a skin rash possibly preceded by drug intake (acetylsalicylic acid, ibuprofen) due to the upper respiratory tract infection. Herpes simplex viral (HSV) infection history was negative. On physical examination she presented diffuse round erythematous macules, some of them creating targetoid lesions, over the neck (Figure 1 B), trunk (Figure 1 C), thighs and elbows along with acral symmetric cellulitis and painful blisters on the dorsal and palmar surfaces (Figure 1 A). Paronychia of almost all fingers was observed. There were small erosions in oral mucosa. The hair and nails were not affected. The patient was obese (BMI 30.5 kg/m2) and diagnosed with mild diabetes 4 months earlier. The family history was negative for dermatological diseases and unremarkable for other conditions.
Figure 1
Cellulitis and blisters on the dorsal surfaces of the feet (A). Erythematous macules and targetoid lesions over the neck (B) and trunk (C)
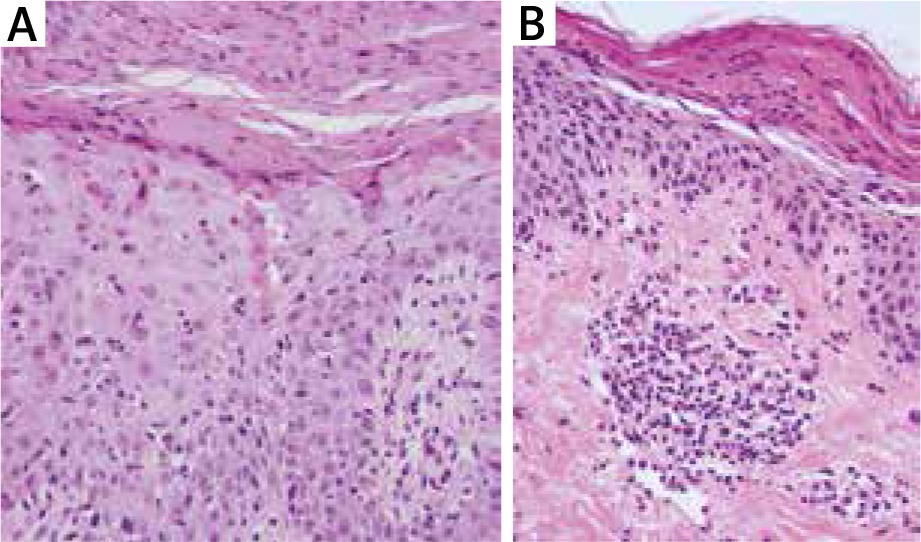
Laboratory investigations revealed lymphocytosis 51.5% (20–40%), hyperglycaemia 6.34 mmol/l (3.9–5.5) and erythrocyte sedimentation rate (ESR) 48 mm/h. Other tests including biochemistry, renal and hepatic profile, thyroid function tests, IgE, ASO were normal. Hepatitis B virus (HBV) and HCV serology were negative. Imaging examinations (chest X-ray and abdominal ultrasonography) were within normal limits. Histopathological examination could be consistent with drug reaction (Figure 2 A). She was diagnosed with erythema multiforme and treated with steroids (dexamethasone intramuscular followed by methylprednisolone per os) and clarithromycin with good response.
Figure 2
Initial biopsy revealed prominent acanthosis and parakeratosis with minimal spongiosis and focal apoptotic eosinophilic keratinocytes (haematoxylin and eosin; 40× magnification) (A). Subsequent biopsy showed irregular acanthosis and prominent parakeratosis (with a partially homogenous parakeratotic layer). In the papillary dermis, focal inflammatory infiltrate composed of lymphocytes, few histiocytes, few granulocytes were observed (haematoxylin and eosin; 40× magnification) (B)
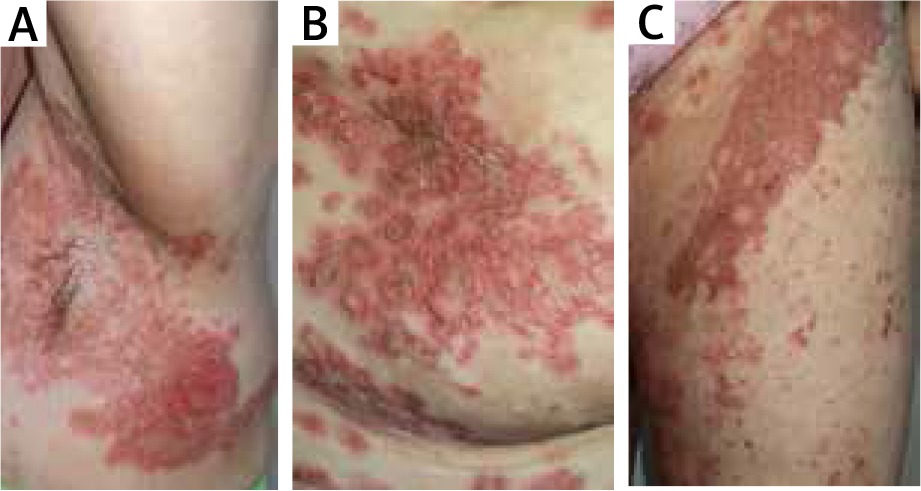
After 2 months the patient was readmitted to our Department due to the worsening of the skin condition. The physical examination revealed erythematous papules and crusted patches predominantly involving the intertriginous regions and scattered over the trunk (Figures 3 A–C). The lesions spread outward leaving postinflammatory hyperpigmentation. The patient reported pruritus and persistent angular cheilitis. Laboratory tests showed an elevated glucose level of 10.49 mmol/l with no other abnormalities. Another skin biopsy was performed and revealed epidermal prominent parakeratosis, mild acanthosis and spongiosis. Perivascular and focal mild infiltration of lymphocytes, histiocytes, neutrophils and eosinophils was observed in the upper dermis (Figure 2 B). The clinical differentials at that time were: psoriasis, pityriasis rubra pilaris, eczema and allergic contact dermatitis. She was treated sequentially with phototherapy (UVB 311 nm) and acitretin 30 mg daily with a rapid exacerbation of skin lesions.
Figure 3
Erosive, erythematous plaques with crusting and scale in the underarm (A), over the trunk (B) and on the groin (C)
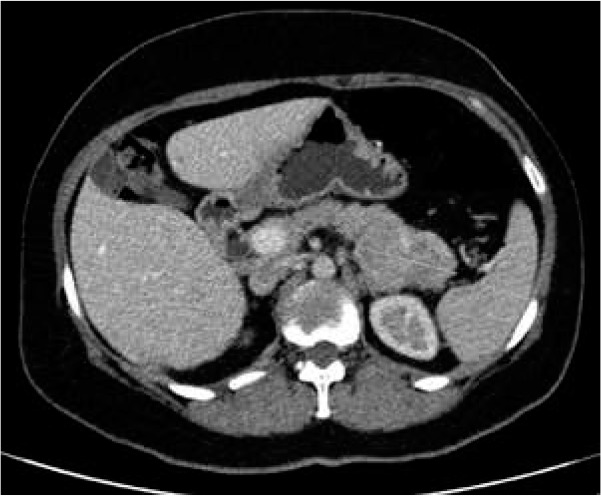
Taking into account an unusual clinical picture and undiagnostic histopathological results, the paraneoplastic syndrome was suspected. The second abdominal ultrasonography disclosed round lesions in the liver – 24 mm in the right lobe and 21 mm in the left lobe. A contrast-enhanced computed tomography of the abdomen confirmed multiple hepatic tumours and low enhancing pancreatic tail mass 56 × 37 mm (Figure 4). The serum glucagon level was highly elevated 2275 ng/l (norm < 209) as well as the levels of chromogranin A 17.2 ng/ml (0–6) and LDH 224 U/l (100–214). She was referred to the Oncology Department where she underwent a needle biopsy of a hepatic lesion that confirmed the neuroendocrine tumour diagnosis (NET G2, Ki-67 3%) with the primary tumour in the pancreas. Afterwards the patient was referred to the Endocrinology Department where she started lanreotide injections of 120 mg once a month. The surgical removal of the tumour was not performed due to the patient’s refusal. For the last 12 months the disease has remained stable.
Figure 4
Computed tomography (CT) of the abdomen showing low enhancing pancreatic tail mass measuring 56 × 37 mm
Glucagonomas are rare neuroendocrine tumours with the incidence rate of 1 in 20 million people with an increasing occurrence in recent decades [4–6]. They can be associated with multiple endocrine neoplasia type 1 (MEN1) in up to 10% but most often are sporadic [7]. They occur mostly in people in their 5th decade of life, equally in males and females, although a case of the paediatric patient was also reported [2, 7].
Glucagon produced by the tumour leads to different symptoms the most characteristic of which is NME present in up to 67% of patients [8]. The clinical presentation involves pruritic erythematous macules that become necrotic or erythematous vesicles and bullae evolving into ulcers and crusted plaques. They affect perineum, perioral and intertriginous areas along with legs, feet and lower abdomen. Koebner phenomenon can be present [9]. Other skin and mucosal changes include paronychia, glossitis, cheilitis and angular stomatitis. Aetiology remains unclear however the catabolic effect of hyperglucagonemia causing hypoaminoacidemia, deficiency of essential fatty acids and zinc are considered as triggering factors. NME is not specific to glucagonoma and can be found in other diseases (cirrhosis, pancreatitis, other malignancies, malabsorption etc.). Other features of glucagonoma include diabetes mellitus (usually mild, controlled with only oral drugs) especially in a patient who has had recent weight loss and normochromic anaemia; diarrhoea, deep vein thrombosis and neuropsychiatric disorders [2, 3, 7]. It is also called 4D syndrome (diabetes, dermatosis, deep vein thromboses and depression) [10].
Histologically presence of epidermal pallor (vacuolated keratinocytes), focal or confluent epidermal necrosis and parakeratosis should suggest the NME although these features are nonspecific and can be absent depending on the stage of evolution of the biopsied lesion [11].
As the clinical appearance and histopathological findings vary patients often remain misdiagnosed as eczema, psoriasis, acrodermatitis enteropathica, zinc deficiency, essential fatty acid deficiency, pellagra, side effects of certain drugs or the initial stages of bullous disease [2]. Patients are waiting for a correct diagnosis for a long time, with median time of 39 months in a case series of 6 patients [12].
By the time of diagnosis tumours usually reach large sizes (0.4–25 cm) and are easily detected in conventional imaging [13]. Computed tomography scans localize tumour mass in the body or tail of pancreas with contrast enhancing due to their hypervascularity [8]. The only curative treatment is complete surgical removal of primary tumour which can be impossible in most cases due to the locally advanced stage and liver metastasis (78% at the time of diagnosis) [6]. For those patients, palliative surgery or chemotherapy may be considered. Even partial resection brings prominent clinical resolution by reducing the glucagon level in the blood [14]. Chemotherapy has low efficacy as the tumours have an extremely low mitotic rate and then chemotherapy is usually replaced by long-acting somatostatin analog (lanreotide, octreotide) – over 80% of glucagonomas express somatostatin receptors [14, 15]. Those agents demonstrate antiproliferative action and reduce glucagon secretion. In the recent study lanreotide was proven to significantly prolong progression-free survival and is recommended for the treatment of patients with gastroenteropancreatic neuroendocrine tumours (grade 1 and 2) (Ki-67 proliferative index < 10%), regardless of the degree of liver involvement and the location of primary tumour [16]. Our patient complied with all of these criteria and lanreotide was used with good initial response and tolerability, which resulted in almost complete resolution of skin lesions.
In conclusion, increased knowledge of polymorphic clinical presentation of necrolytic migratory erythema can avoid delay in the recognition. We present a case with rapid onset of erythema multiforme-like eruptions which evolved over a few months into characteristic features of NME facilitating the early diagnosis.







