
Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, is a rare, idiopathic, non-Langerhans cell histiocytic disorder first described by Rosai and Dorfman in 1969 [1]. It is characterized by the proliferation of histiocytes in lymph nodes and various other tissues. Although the disease typically presents with painless, massive, bilateral lymphadenopathy, it can also manifest at extranodal sites, making it a diagnostic challenge [2]. We present the case of an adult patient with RDD who initially presented with angioedema, an unusual symptom of this rare condition.
A 62-year-old Caucasian woman presented to the Dermatology and Allergy Department with a 5-year history of recurrent oedema episodes. The oedema was localized to the lower face and neck. The patient also had swallowing and breathing problems. Erythema and oedema also appeared around her eyes (Figure 1). The patient experienced chronic fatigue, excessive sweating, and vertigo. The standard urticarial treatment was ineffective. Hereditary angioedema was excluded. Lymph nodes located in the neck were palpable. Positron emission tomography confirmed proliferative enlargement of the cervical lymph nodes, and biopsy was performed. Histological imaging revealed an enlarged node with sinusoidal expansion and diffuse infiltration of histiocytes (CD163+; CD1a-, S-100+). Lymphoma was excluded. The image was indicative of RDD (Figure 2). The enlarged lymph nodes were further evaluated using immunohistochemistry, which revealed positive staining for CD163 and S-100, but negative staining for CD1a, supporting the diagnosis of RDD. The course of the disease was stable. The patient received prednisolone in the dose of 10 mg/day and methotrexate of 20 mg per week with folic acid of 15 mg per week, with a good clinical response.
Figure 1
Periorbital and lower facial oedema in a 62-yearold woman with RDD. Erythema and swelling were recurrent and unresponsive to standard antihistamine therapy, initially mimicking angioedema of other aetiologies
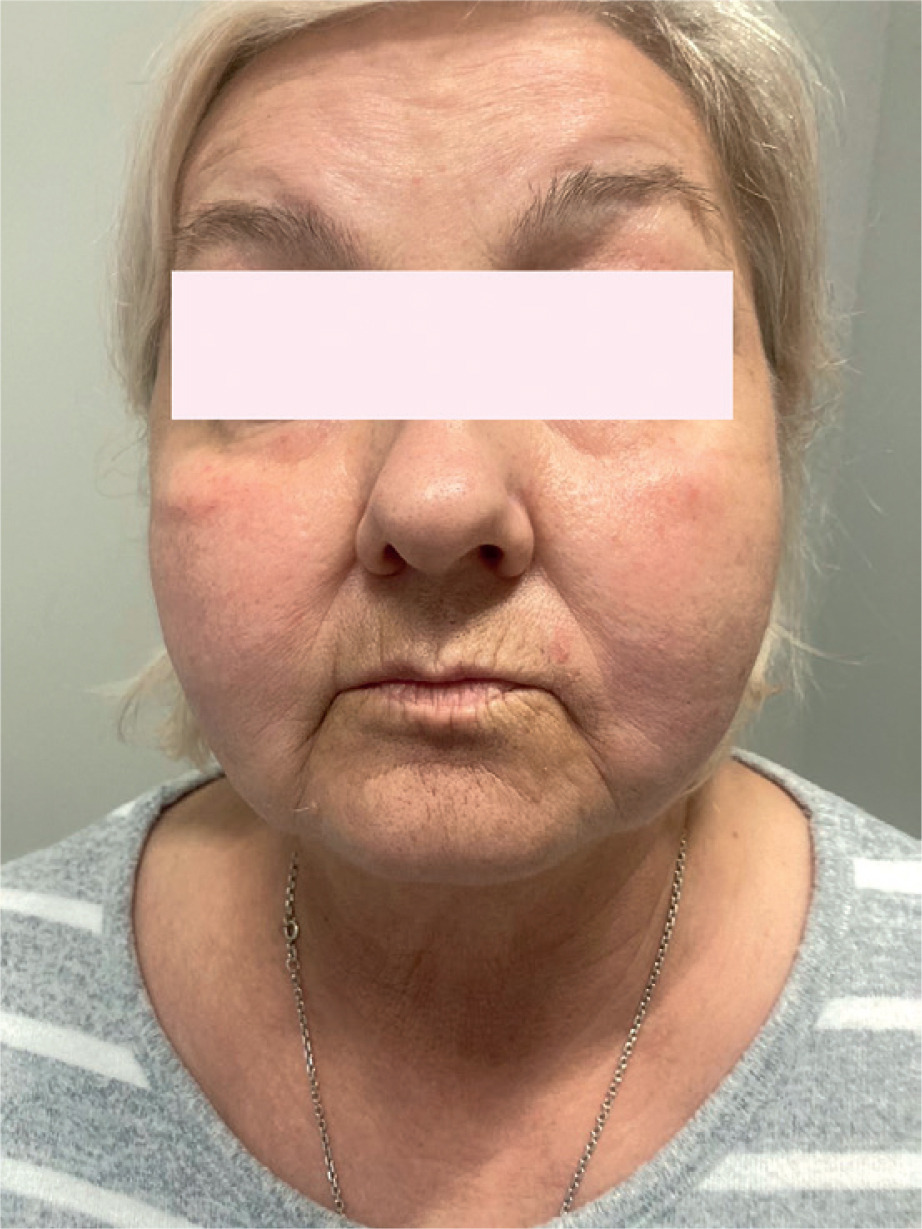
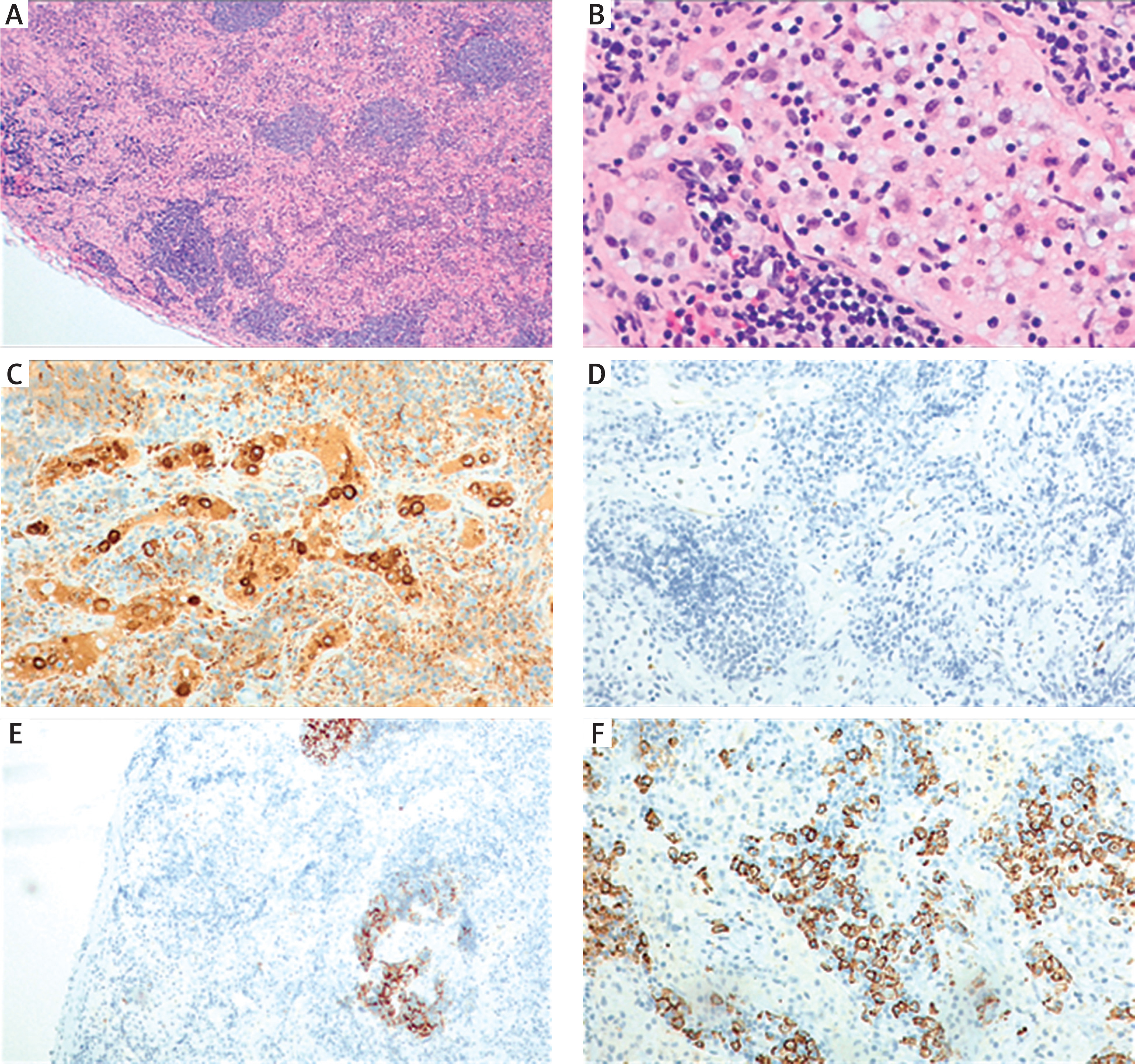
Figure 2
Histopathological image of a lymph node from a patient with Rosai-Dorfman disease. A, B – Haematoxylin & eosinstained sections showing distorted nodal architecture with expanded interfollicular zones containing numerous histiocytes with pale cytoplasm and emperipolesis (magnifications 40× and 400×, respectively). C, D – Immunohistochemistry highlighting large histiocytes with pale cytoplasm and emperipolesis (C – CD68-positive; D – CD1a-negative; both 200×). E – CD23 staining demonstrates a damaged but preserved follicular dendritic cell (FDC) meshwork within residual small follicles (100×). F – Marked plasmacytosis in CD138 staining in the interfollicular area (200×)
RDD is a rare idiopathic disorder that can present with a wide range of clinical manifestations, including extranodal involvement. In this case, the patient presented with angioedema, an unusual initial symptom. Characteristic histopathological findings, such as the presence of large histiocytes exhibiting emperipolesis (engulfment of lymphocytes, erythrocytes, or other cells), as well as a specific immunohistochemical profile (positive for CD163 and S-100, negative for CD1a), are essential for the diagnosis [3]. The diagnosis of RDD can be challenging because the disease can mimic various other conditions such as malignancies, infections, and autoimmune disorders. The presence of angioedema as an initial symptom highlights the importance of considering RDD in the differential diagnosis of atypical swelling or inflammation. The early recognition and diagnosis of RDD can lead to timely and appropriate management strategies, potentially improving patient outcomes. The pathogenesis of RDD is not well understood, but it is thought to involve a dysregulated immune response that leads to histiocyte proliferation [1]. The treatment of RDD is highly individualized, depending on the extent of the disease, symptomatology, and molecular profile. While localized disease can often be managed with observation or surgical excision, systemic or refractory disease may require a combination of corticosteroids, chemotherapy, and targeted therapies. Ongoing research into the molecular pathogenesis of RDD is expected to further refine treatment strategies, particularly with the advent of targeted therapies [4–6].
In conclusion, RDD is a rare, idiopathic disorder that can present with a wide range of clinical manifestations, including extranodal involvement. Clinicians should be aware of the potential for atypical presentations such as angioedema to ensure timely diagnosis and appropriate management of this rare condition. The rarity of RRDD underscores the importance of maintaining a high index of suspicion, especially when unusual clinical presentations are encountered [7–9]. Increased awareness among health care professionals can lead to improved recognition and diagnosis of this condition, potentially reducing delays in treatment initiation. While the underlying aetiology of RDD remains incompletely understood, ongoing research has provided insights into the potential role of immune dysregulation and genetic alterations involving the MAPK/ERK pathway [5]. Further investigation into the pathogenic mechanisms and development of targeted therapeutic approaches may lead to improved management strategies for this rare and challenging condition.







